
团簇Fe3Ni3电子性质
2021-10-13 23:06郑新喜方志刚秦渝侯欠欠吴庭慧毛智龙
贵州大学学报(自然科学版) 2021年5期
郑新喜 方志刚 秦渝 侯欠欠 吴庭慧 毛智龙
摘 要:为探究团簇Fe3Ni3优化构型的电子性质,使用密度泛函理论中的B3LYP/Lanl2dz (Level)对设计出的初始构型进行全参数优化计算,排除含虚频和能量较高的相同构型后,最终得到9种稳定的优化构型。从各优化构型的电荷量、原子内轨道布居数、原子及原子间的自旋布居数、原子自旋密度分析发现:团簇Fe3Ni3内部Fe原子一般为电子供体,Ni原子一般作为电子受体;构型内部各原子的4s轨道是电子流出的主要贡献者,3d轨道是电子流入的主要贡献者,且4p轨道对电子流入的贡献不可忽视;三重态构型内部β电子自旋密度越大,α和β2种电子云的重叠程度越高,构型的稳定性越好,其中构型1(3)与构型2(3)的稳定性最好,构型5(3)稳定性最差。
关键词:团簇Fe3Ni3;密度泛函理论;电子性质;电子自旋密度
中图分类号:O641.12
文献标志码:A
随着2020年《区域全面经济伙伴关系协定》(regional comprehensive economic partnership,RCEP)协议签订,为我国能源短缺问题带来了新的机遇,其中新能源氢能具有非常好的发展前景。目前,电解水为制备氢能的主要方法。传统铂碳催化剂虽具有良好的催化性能,但由于铂金属价格昂贵,很大程度上限制了其大规模工业化应用,因此急需寻找一种新型催化材料。
微观结构具有长程无序、短程有序等特征的非晶态合金,与传统的晶态合金相比,这种新材料在催化性能[1-2]、储氢性能[3-4]、磁性[5-6]等方面展现出优异性能,自发现以来,便受到众多学者深入的研究并广泛应用于众多领域。其中Fe基[7]、Ni基[8-9]由于成本低廉、环保无毒、物理化学性能优越等因素,已成为非晶态合金相关研究的热点,例如Fe-Cr[10-11]、Fe-Co[12-13]、Fe-Cr-Ni[14-15]等体系均已取得众多显著的研究成果。其中,非晶态Fe-Ni合金在纳米C纤维结构中的电解水催化析氧性能[16-17]表现突出,成为电解水催化剂方面的新型材料,其优异的催化性能为能源短缺问题提供了一种新的解决思路。尽管非晶态Fe-Ni合金有众多优异性能与良好的发展前景,但其催化的微观理论研究目前鲜有报道,不能很好地对其宏观性质起到理论支撑作用。同时,YANG等[18]的研究成果表明,团簇Fe-Ni在原子比例为1∶ 1时表现出最佳的催化性能。因此,本文设计了团簇Fe3Ni3结构模型,并从电子性质与催化活性两个方面对团簇Fe3Ni3展开研究。
1 优化构型及能量
依据拓扑学原理[29],将团簇Fe3Ni3所有可能存在的典型构型设计出来,共得到19种初始构型。使用密度泛函理论[20-21](density functional theory, DFT)中的B3LYP/Lan12dz (Level),对团簇Fe3Ni3的19种初始构型分别在单、三重态下进行全参数优化计算。其中,Fe、Ni原子采用WADT等[22]的含相对论校正的有效核电势价电子从头计算基组,即18-eECP的双ξ基组(3s,3p,3d/2s,2p,2d)。所有运算均在计算机Z440上运用Gaussian 09程序[23]完成。
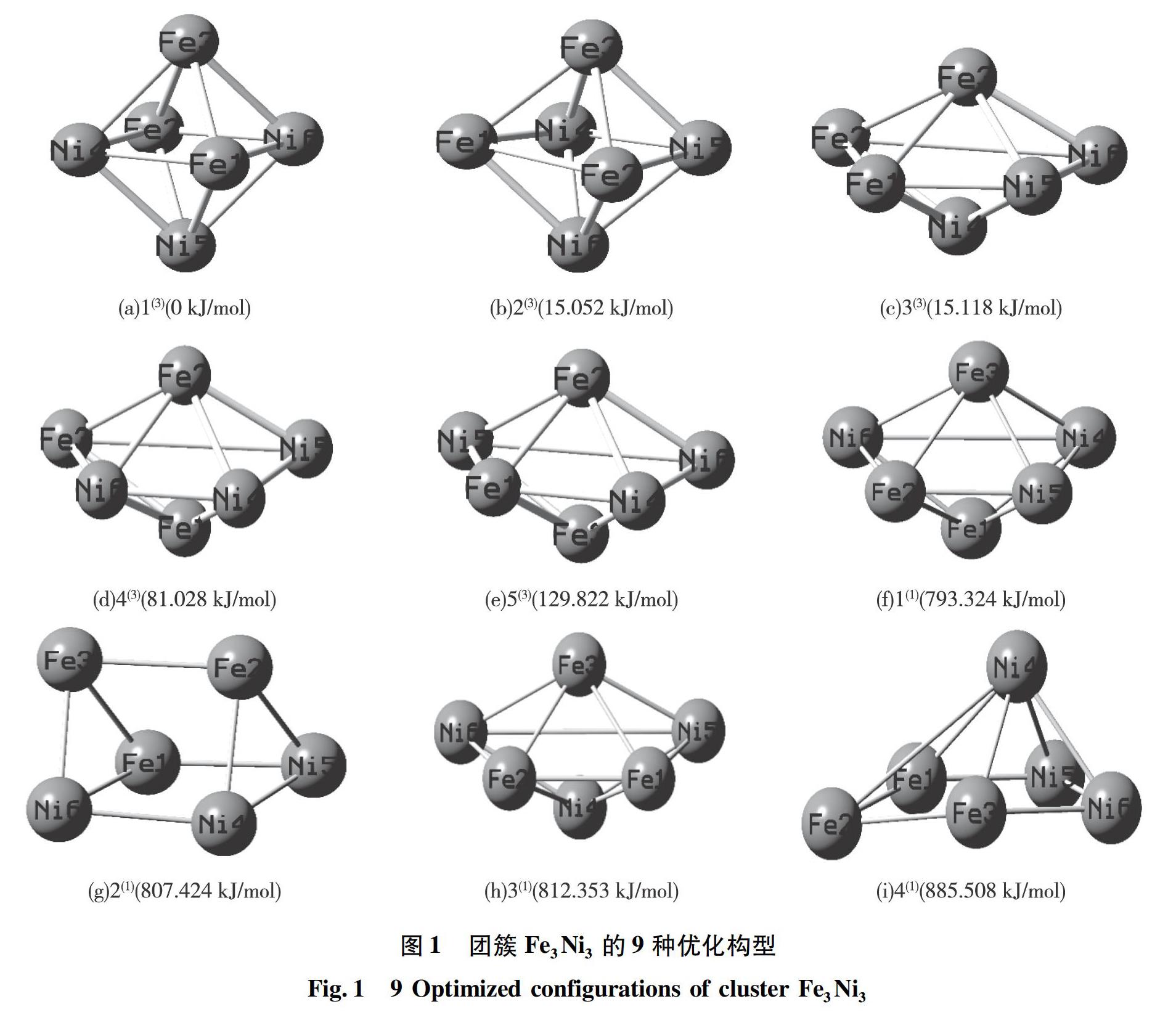
对优化计算后的数据进行处理,排除不稳定的含虚频构型和同重态下能量较高的相同构型后,最终得到9种优化构型,其中,三重态构型5种,单重态构型4种。
将具有最低校正能的构型1(3)作为基准(0 kJ/mol),计算出其他8种构型的相对能量并标注于各构型下方,且序号上标括号内的数字为构型的自旋重态,如图1所示。将9种优化构型按照热力学稳定性(即校正能量的大小)由高到低排序:1(3) >2(3) >3(3) >4(3) >5(3) >1(1) >2(1) >3(1) >4(1)。由排序发现三重态优化构型的能量均小于单重态,说明单重态各优化构型的稳定性较差,且构型1(3)的稳定性最好,构型4(1)的稳定性最差。通过观察优化构型的空间立体图可以看出,团簇Fe3Ni3的三重态优化构型均为四角双锥型,而单重态中存在四角双锥型(1(1)和3(1))、三棱柱型(2(1))和五棱锥型(4(1))。
2 团簇Fe3Ni3的电子性质
2.1 团簇Fe3Ni3各原子电荷量
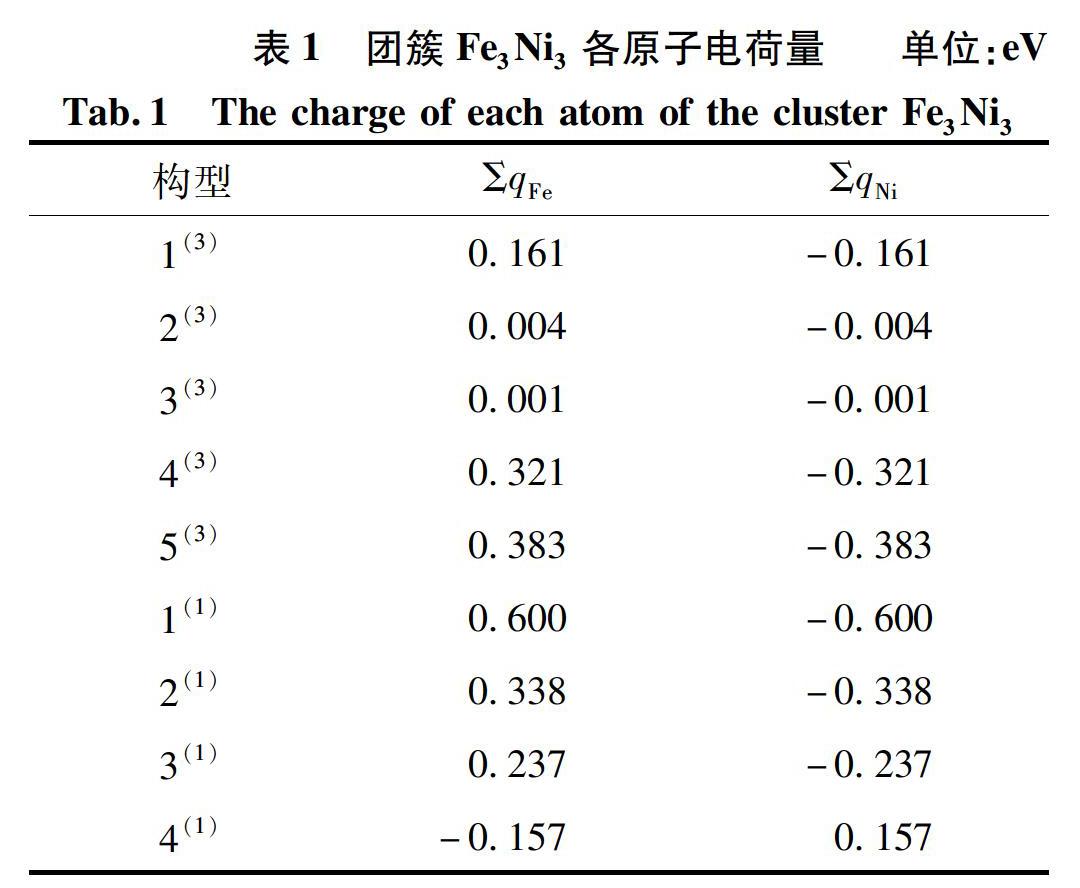
原子电荷量是一种可以用来表征团簇构型内部电子性质的重要参数,对其具体分析可以探究团簇Fe3Ni3的电子流向和电子流动性强弱。原子电荷量为正值表示电子从该原子流出,电荷量为负值则表征为电子流入该原子,且电荷量绝对值的大小能够直接反应电子流动性的强弱。因此,为便于探究团簇Fe3Ni3的电子性質,将9种优化构型中2种原子的总电荷量数据列于表1。
由表1可以明显看出:团簇Fe3Ni3的9种优化构型中,Fe、Ni 2种原子的总电荷量之和均为0,说明该团簇所有优化构型均呈电中性,可以在现实中稳定存在。其中,除构型4(1)以外的所有构型中,Fe原子电荷量总和均为正值,Ni原子电荷量总和均为负值。由此说明:在团簇Fe3Ni3中除构型4(1)以外的其他构型,Fe原子是电子的提供者,而Ni是电子的接受者,即团簇内电子流动方向为Fe原子→Ni原子;而构型4(1)的内部电子流动方向为Ni原子→Fe原子。对其空间结构进行研究发现,构型4(1)与其他8种构型空间结构均不同,说明构型的空间结构对电子流向具有一定的影响。
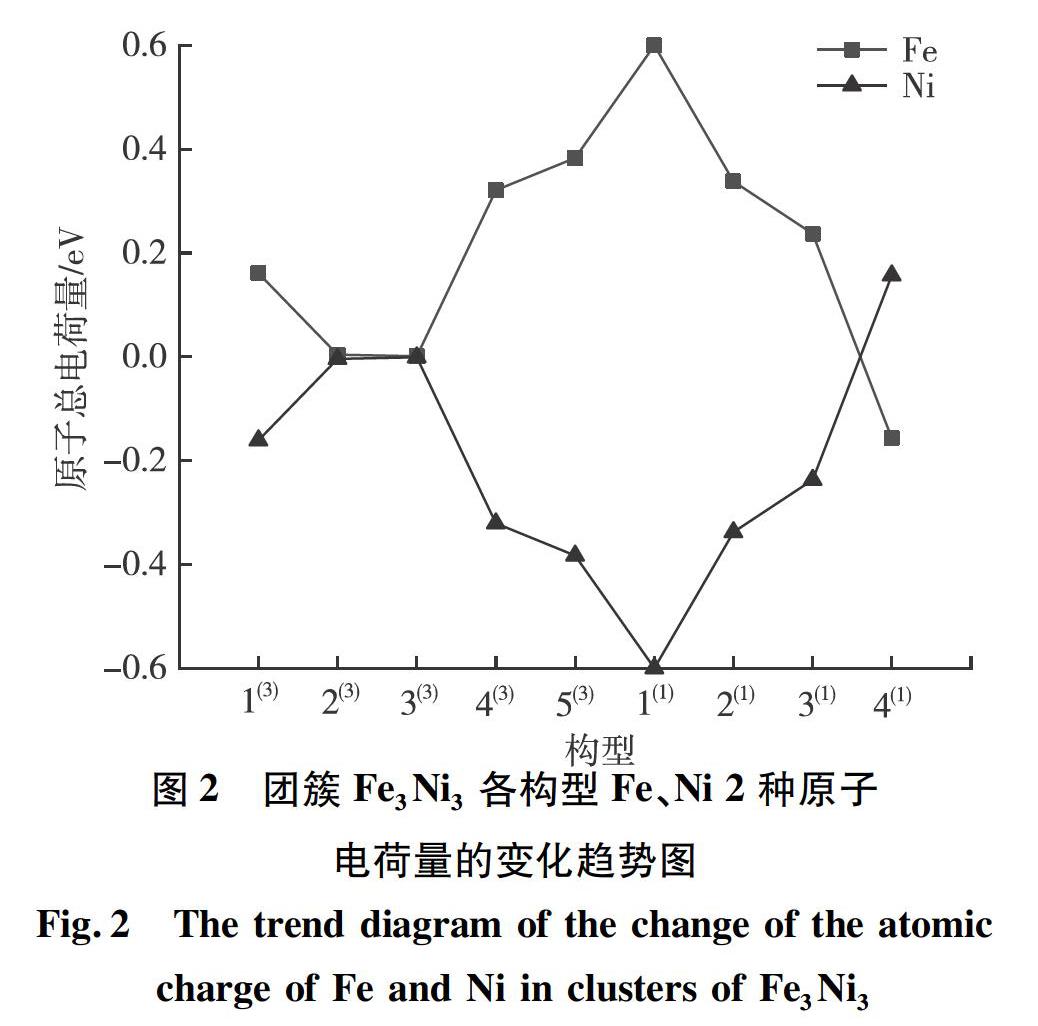
为更加直观地探究团簇Fe3Ni3各优化构型的电子流动性强弱,由表1数据绘制出团簇Fe3Ni3中Fe、Ni 2种原子电荷量的变化趋势,如图2所示。由于该团簇仅有2种原子,且正负电荷量相互抵消,故2种原子电荷量变化趋势相反,对其一进行分析即可得出各构型电子流动性强弱关系。本文选取对Fe原子的电荷量进行分析。由图2可知:团簇Fe3Ni3中构型1(1)的Fe原子总电荷量最大,说明该构型内部电子流动性最强;而构型2(3)与构型3(3)的Fe原子总电荷量很小(构型2(3)是0.004、构型3(3)是0.001),几乎为零,说明这2种构型内电子流动性最弱。因此,团簇Fe3Ni3优化构型的电子流动性强弱关系:1(1) >5(3) >2(1) >4(3) >3(1) >1(3) >4(1) >2(3)≈3(3)。
2.2 团簇Fe3Ni3各原子轨道布居数变化分析
微观分子化学中,将分子的电子云密度转化为由分子中各原子不同轨道的电子组成,而电荷在各个原子轨道都有“布居”的存在,用布居数表示。对其进行分析,可以探究电荷在原子各轨道上分布差异以及电子转移等现象。布居数数值为正值时,电子流入该轨道;布居数数值为负值时,电子从该轨道流出。对团簇Fe3Ni3各优化构型布居数进行分析,见表2。
由表2可以看出,Fe、Ni 2种原子的4s轨道布居数均为负值,而3d、4p轨道布居数均为正值,说明各构型内部电子由s軌道流向p、d轨道。将各原子d、p轨道布居数作进一步比较发现,d轨道的布居数均大于p轨道布居数,说明d轨道得电子能力大于p轨道得电子能力。在团簇Fe3Ni3各优化构型中,Fe、Ni的4s轨道是主要电子供体,3d轨道是主要电子受体,但各原子的4p轨道对电子流入的贡献同样不可忽视。对团簇2种原子的布居数求和可知,Ni原子的布居数除构型4(1)外,其余构型均为正值,说明Ni原子具有较强的得电子能力,这也与前文2.1中“电子流向为:Fe原子→Ni原子”的结论相互印证。
2.3 团簇Fe3Ni3的原子及原子间的自旋布居分析
2.3.1 团簇Fe3Ni3各原子的自旋布居分析
原子的自旋布居数可以用来表征团簇Fe3Ni3的电子自旋情况。因此,对团簇Fe3Ni3各原子的电子自旋布居进行分析,可以探究出团簇各原子电子自旋情况。采用Multiwfn [24]对各个原子周围的电子自旋密度进行全空间积分,所得的结果为各原子周围的自旋布居数,见表3。自旋布居数为正值表示自旋向上的α单电子出现的净概率密度更大,自旋布居数为负值表示自旋向下的β单电子出现的净概率密度更大。由于单重态构型为闭壳层,其2个自旋的空间轨道同等,基态分子的电子自旋成对,即净自旋为零,研究并无实际意义,故此部分仅对团簇Fe3Ni3的三重态构型进行研究。
由表3可知:构型1(3)—3(3)中均存在2个Fe原子与1个Ni原子呈自旋向上的α电子、2个Ni原子和1个Fe原子为自旋向下的β电子;而构型4(3)与构型5(3)均存在2个Fe原子带自旋向下的β电子、一个Fe原子和3个Ni原子带自旋向上的α电子;同时各构型Fe原子的电子自旋布居数绝对值均大于Ni原子,说明Fe原子的成单电子数大于Ni原子,且各构型中Fe原子与Ni原子的总电子自旋布居数基本相反。
2.3.2 团簇Fe3Ni3各原子间的自旋布居分析
若仅对团簇各原子的电子自旋布居进行分析,有一定的局限性。因此,为更加全面、深入地了解团簇Fe3Ni3电子自旋情况,继续对构型各原子间电子自旋布居进行探究。原子间的电子自旋布居是判断构型原子间成键强度的一个重要依据。电子自旋布居数的绝对值可以反映原子间成键强度的强弱,进而推断构型成键情况。团簇Fe3Ni3各原子间电子自旋布居数具体数据见表4。其中,数值为正表示两原子间成键时α电子盈余,数值为负则表示两原子间成键时β电子盈余。
由表4可以看出:在最稳定的构型1(3)中,Fe—Fe键与Fe—Ni键的电子自旋布居数均为负值,说明这2种成键方式的电子盈余为β电子;在稳定性最差的构型5(3)中,Fe—Fe键、Fe—Ni键与Ni—Ni键的电子自旋布居数均为正值,说明该构型中3种成键方式的电子盈余均为α电子。将三重态的5种构型分为3组:稳定性高的A组(构型1(3)与2(3))、稳定性较高的B组(构型3(3)与4(3))与稳定性较差的C组(构型5(3))。分析数据发现:A组中有2种成键方式的电子自旋布居数为负值,成键时为β电子盈余;B组有1种成键方式的电子自旋布居数为负值,成键时为β电子盈余;C组无负值,说明原子间成键β电子盈余时有助于构型的稳定性。
2.4 团簇Fe3Ni3的电子自旋密度图
电子自旋密度图可以用来表征团簇Fe3Ni3构型内部α电子与β电子在原子和原子间的重叠分布情况,进而分析构型的稳定性,如图3所示。图中浅白色表示α电子,深黑色表示β电子。由图3可知:A组(构型1(3)与2(3))的电子自旋密度图,深黑色的电子云大于浅白色的电子云,说明该构型内部β电子较多,且2种电子的电子云重叠度较高,构型稳定性较高;稳定性较好的B组(构型3(3)与4(3))的2种电子云重叠程度较差,且明显发现构型4(3)的浅白色α电子云远大于深黑色的β电子,且β电子基本被α电子包围;稳定性差的C组(构型5(3))的β电子云较少,且与α电子云的重叠程度最差。由此说明,团簇Fe3Ni3构型内部β电子云越大,以及α和β 2种电子云的重叠程度越高,构型的稳定性就越好。
3 结论
本文从微观电子流动性方面分析了团簇Fe3Ni3各构型的稳定性,主要涉及了电荷、原子各轨道布居数、原子及原子间自旋布居数、自旋密度图等相关因素。
1)团簇Fe3Ni3共有9种优化稳定构型,其中,三重态构型5种,单重态构型4种,三重态构型稳定性均优于单重态构型。
2)团簇Fe3Ni3除构型4(1)以外,构型内电子流动方向为:Fe原子→Ni原子;各构型内部电子由4s轨道流向4p、3d轨道,4s轨道是电子流出的主要贡献者,且3d轨道得电子能力大于4p轨道,是电子流入的主要贡献者,但各原子的4p轨道对电子流入的贡献不可忽视。
3)团簇Fe3Ni3三重态构型内部β电子自旋密度越大,α和β 2种电子云的重叠程度越高,构型的稳定性越好。其中,构型1(3)与构型2(3)的稳定性最好,构型5(3)稳定性最差。
参考文献:
[1]GLASSCOTT M W, PENDERGAST A D, Goines S, et al. Electrosynthesis of high-entropy metallic glass nanoparticles for designer, multi-functional electrocatalysis[J]. Nature Communications, 2019, 10(1): 1-8.
[2]ZUO L, LI R, JIN Y, et al. Homogeneous nanoporous Ni particles produced by dealloying Mg-based metallic glass as efficient hydrogen evolution electrocatalyst[J]. Journal of The Electrochemical Society, 2018, 165(3): 207-214.
[3]RVSZ A, KIS-TTH A, SZOMMER P, et al. Hydrogen storage, microstructure and mechanical properties of strained Mg65Ni20Cu5Y10 metallic glass[J]. Materials Science Forum, 2013, 729: 74-79.
[4]秦渝, 方志剛, 张伟, 等. Co3NiB催化析氢活性研究[J]. 江西师范大学学报(自然科学版), 2020, 44(1): 56-62.
[5]徐诗浩, 方志刚, 韩建铭, 等. 团簇V3B2成键及磁学性质研究[J]. 广西师范大学学报(自然科学版), 2017, 35(3): 89-96.
[6]LYU Y B, CHEN Q J, HUANG Y L. Tunable Curie temperature and magnetocaloric effect of FeCrMoCBYNi bulk metallic glass with different crystallized phases[J]. Journal of Rare Earths, 2019, 37(4): 74-79.
[7]杜浩, 董俊. Fe(3.84)Ni(5.57)B(0.69)非晶合金的非等温晶化动力学研究[J]. 贵州大学学报(自然科学版), 2009, 26(3): 28-30, 63.
[8]李历红, 方志刚, 赵振宁, 等. 团簇Ni3CoP电子性质与磁性研究[J]. 江西师范大学学报(自然科学版), 2019, 43(2): 160-166.
[9]WU W Z, JIANG J L, LI G W, et al. Ultrasonic additive manufacturing of bulk Ni-based metallic glass[J]. Journal of Non-Crystalline Solids, 2019, 506: 1-5.
[10]ZHANG B, WANG J, WU B, et al. Unmasking chloride attack on the passive film of metals[J]. Nature Communications, 2018, 9(1): 1-9.
[11]廖薇, 方志刚, 赵振宁, 等. 团簇Fe3Cr3的电子性质与成键[J]. 辽宁科技大学学报, 2019, 42(2): 101-106.
[12]LIU J Y, LI X N, RYKOV A I, et al. Zinc-modulated Fe-Coprussian blue analogues with well-controlledmorphlogy for efficient cesium sorption[J]. Journal of Materials Chemistry A, 2017, 5(7): 3284-3292.
[13]YAMAZAKI T, YAMAMOTO T, FURUYA Y, et al. Magnetic and magnetostrictive properties in heat-treated Fe-Co wire for smart material/device[J]. Mechanical En-gineering Journal, 2018, 5(2): 1-9.
[14]TIAN Y, GORBATOA O I, BORGENSTAM A, et al. Deformation microstructure and deformation-inducedmartensite in austenitic Fe-Cr-Ni Alloys depending on stacking fault energy[J]. Metallurgical & Materials Transactions A, 2017, 48(1): 1-7.
[15]KUMAR A, KHATIRKAR R K, GUPTA A, et al.Deci-phering the possible role of strain path on the evolution of microstructure, texture, and magnetic properties in a Fe-Cr-Ni alloy[J]. Metallurgical & Materials Transac-tions A, 2018, 49(8): 3402-3418.
[16]TAO X, XU H, LUO S, et al. Construction of N-doped carbon nanotube encapsulated active nanoparticles in hierarchically porous carbonized wood frameworks to boost the oxygen evolution reaction[J]. Applied Catalysis B: Environmental, 2020, 279: 119367.1-119367.9.
[17]ZHANG Y N, HOU X B, LI X, et al. FeNi Alloy nanoparticles embedded in electrospun nitrogen-doped carbon fibers for efficient oxygen evolution reaction[J]. Journal of Colloid and Interface Science, 2020, 578: 805-813.
[18]YANG L, ZENG X F, WANG D, et al. Biomass-derived FeNi alloy and nitrogen-codoped porous carbons as highly efficient oxygen reduction and evolution bifunctional electrocatalysts for rechargeable Zn-air battery[J]. Energy Storage Materials, 2018, 12: 277-283.
[19]YU X G. Hyperbolic multi-topology and the basic principle in quantum mechanics[J]. Advances in Applied Clifford lgebras, 1999, 9(1): 109-118.
[20]BECK A. Density-functional thermochemistry. III. Therole of exact exchange[J]. The Journal of Chemical Physics, 1993, 98(7): 5648-5652.
[21]李雯博, 方志刚, 赵振宁, 等. 团簇Co5B2反应活性的DFT研究[J]. 廣西师范大学学报(自然科学版), 2017, 35(4): 76-83.
[22]WADT W R, HAY P J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi[J]. The Journal of Chemical Physics, 1985, 82(1): 284-298.
[23]FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09, Revision C.01[CP/DK]. Wallingford CT: Gaussian, Inc, 2010.
[24]LU T, CHEN F W. Multiwfn: a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592.
(责任编辑:周晓南)
Abstract:
In order to explore the electronic properties of the optimized configuration of Fe3Ni3 clusters, the B3LYP/Lanl2dz (Level) in density functional theory was used to perform full-parameter optimization calculations on the designed initial configuration, and the same configuration with false frequency and higher energy was excluded. After forming, 9 stable optimized configurations are finally obtained. From the analysis of the charge amount of each optimized configuration, the number of orbital populations in the atom, the number of spin populations between atoms and between atoms, and the atomic spin density analysis, it is found that: the Fe atoms in the cluster Fe3Ni3 are generally electron donors, and Ni atoms are generally used as electron acceptors; the 4s orbitals of each atom in the configuration are the main contributors to the outflow of electrons, and the 3d orbitals are the main contributors to the inflow of electrons. The contribution of the inflow of electrons cannot be ignored; the greater the β electron spin density in the triplet configuration, the higher the overlap of the α and β electron clouds, and the better the stability of the configuration. Among them, configuration 1(3) and configuration 2(3) have the best stability, and configuration 5(3) has the worst stability.
Key words:
cluster Fe3Ni3; density functional theory; electronic properties; electron spin density
