
G 蛋白耦联受体激酶2 参与炎症痛的研究进展
2021-10-13 08:50:28李晓晨综述王彦青毛应启梁审校
复旦学报(医学版) 2021年5期
李晓晨(综述) 陈 愈 王彦青 毛应启梁(审校)
(复旦大学基础医学院中西医结合学系 上海 200032)
炎症痛是一种临床常见的疼痛类型。全球大约有两千万例类风湿性关节炎患者,从1990 年至2017 年增加了 7.4%[1]。2017 年全球大约有 3 亿骨关节炎患者[2],我国膝关节症状性骨关节炎的患病率为8.1%[3],美国关节炎患者中约有四分之一伴有严重的关节疼痛[4]。美国20~69 岁成年人中炎症性背痛的患病率大约为5%~6%[5]。目前药物治疗以非甾体类抗炎药、阿片类药物为主,但非甾体类抗炎药往往具有胃肠和心血管等方面的副作用,并且不能有效治疗较高强度的疼痛,故阿片类镇痛药由于其副作用、耐受性和依赖性,临床应用受到一定的限制。G 蛋白耦联受体激酶2(G-protein-coupled receptor kinase 2,GRK2)广泛表达于各种组织中,已有研究表明GRK2 在急性炎症痛转为慢性炎症痛的过程中发挥重要作用,脊髓或DRG 神经元内过表达GRK2 可以缓解慢性炎症痛。目前尚无这方面的中文综述,故本文就近年来GRK2 在炎症痛中的作用研究进行总结,以期为炎症痛的机制研究及新药开发提供参考。
GRK2 的结构、分布及功能GRK2 最初被称为β-肾上腺素能受体激酶1(β-adrenergic receptor kinase 1,βARK-1),是 GRKs 家 族 成 员 之 一[6]。GRK2 的结构类似于其他GRKs,由3 个主要的结构域组成:中央激酶结构域、氨基末端区域和羧基末端区域。氨基末端区域旁有G 蛋白信号调节区域[7],羧基末端区域有 Pleckstrin Homolgy(PH)结构域( 图 1)。 磷 脂 酰 肌 醇 4,5- 磷 酸 氢 盐(phosphatidylinisitol 4,5-bisphosphate,PIP2)和游离的 Gβγ 亚基通过与 GRK2 的 PH 结构域相互作用,使胞质GRK2 转位到细胞膜上,从而发挥激酶调控作用[6-9]。GRK2 在哺乳动物心脏、血管内皮细胞、脑、肾近曲小管、肺、肝脏和骨骼肌等组织中均有表达,在神经和免疫系统中高水平表达[10-11]。在脊髓中,GRK2 在背角浅层表达水平相对较高[12]。在脑内,GRK2 mRNA 几乎均匀地分布于皮层各层,在丘脑、海马、黑质致密区、腹侧被盖区和蓝斑等核团也都有分布[13]。
GRK2 的主要功能是调节激动剂诱导的G 蛋白耦联受体快速脱敏及信号转导,保护细胞免受过度刺激。 G 蛋白耦联受体(G-protein-coupled receptors,GPCRs)是最大的信号蛋白家族,许多参与疼痛的神经递质通过GPCRs 传递信号。外周伤害性感受器上的GPCRs 能够感知包括前列腺素、组胺、腺苷等在内的多种激活分子,促使初级传入神经元的中枢末端释放谷氨酸、P 物质和降钙素基因相关肽(calcitonin gene related peptide,CGRP)等神经递质,激活脊髓背角投射神经元上的GPCRs,使得疼痛信号得以传递至大脑[14]。有证据表明,痛觉过敏和机械性痛觉超敏与GPCRs 的敏感性增加有关,如沙鼠坐骨神经慢性压迫性损伤(chronic constriction injury,CCI)模型足底注射 NK-1 受体激动剂可以增强痛觉过敏[15]。而当GPCRs 被激活时,GRK2 能够磷酸化GPCRs 的丝氨酸/苏氨酸位点,导致GPCRs 与G 蛋白解耦联,招募抑制蛋白βarrestin 与 GPCRs 结 合 ,从而导致 GPCRs 的 快 速 脱敏[16-17](图 1)。GRK2 水平的改变可以调节与疼痛相关的GPCRs 的反应性,GRK2 水平升高促进一系列GPCRs 脱敏,使得细胞对激动剂的反应降低,相反,GRK2 水平降低导致 GPCRs 激活延长[12]。

图1 GRK2 的结构与GRK2 介导的受体脱敏Fig 1 The structure of GRK2 and GRK2-mediated receptor desensitization
除此之外,GRK2 还可以直接和细胞内信号分子如丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族成员、磷脂酰肌醇 3-激酶(phosphatidylinositol 3-kinase,PI3K)信号通路相关分子、细胞骨架蛋白相互作用[11]。有研究表明,GRK2 通过丝裂原活化蛋白激酶激酶(mitogenactivated protein kinase kinase,MEK)/细胞外信号调 节 激 酶(extracellular signal-regulated kinase,ERK)信号通路调控C-C 趋化因子受体2(C-C motif chemokine receptor 2,CCR2)诱导的 ERK 激活[18]。GRK2 通过和 PI3K 形成胞质复合物参与激动剂诱导的 PI3K 向细胞膜募集的过程[19];另外,已有研究证明GRK2 可以结合并磷酸化几种非受体底物,包括突触核蛋白、光导蛋白、埃兹蛋白和微管蛋白[10]。这使得 GRK2 可以不依赖于 GPCRs,直接调节细胞内信号传导[20]。
GRK2 与炎症痛已有临床研究表明,与健康对照相比,类风湿性关节炎或多发性硬化症患者外周血单核细胞的GRK2 蛋白水平降低;同样在类风湿性关节炎或多发性硬化的动物模型中,脾细胞GRK2 蛋白水平也降低[21]。在基础研究中,越来越多的研究表明GRK2 参与了急性炎症痛转为慢性的过程。与对照组相比,初级感觉神经元和脊髓小胶质细胞内 GRK2 都显著降低[20,22]。在角叉菜胶、C-C趋化因子配体 3(C-C motif chemokine ligand 3,CCL3)、白介素 1β(interleukin-1β,IL-1β)、肾上腺素(epinephrine,EPI)、前列腺素 E2(prostaglandin E2,PGE2)诱导的炎症痛模型中,GRK2+/-小鼠的痛觉过敏可延长至 10~21 天[20,22-24]。
有证据表明,在小胶质细胞/巨噬细胞/粒细胞内特异性敲减GRK2(LysM-GRK2+/-小鼠)能够延长角叉菜胶引起的痛觉过敏至3 周,但是在伤害性感觉神经元特异性敲减GRK2(SNS-GRK2+/-小鼠)仅延长角叉菜胶引起的痛觉过敏至8 天左右[11]。以上结果提示不同神经细胞中的GRK2 参与炎症痛的机制可能不同。下面将分别讨论神经元、小胶质细胞和星形胶质细胞中的GRK2 在炎症痛中的作用(图2)。
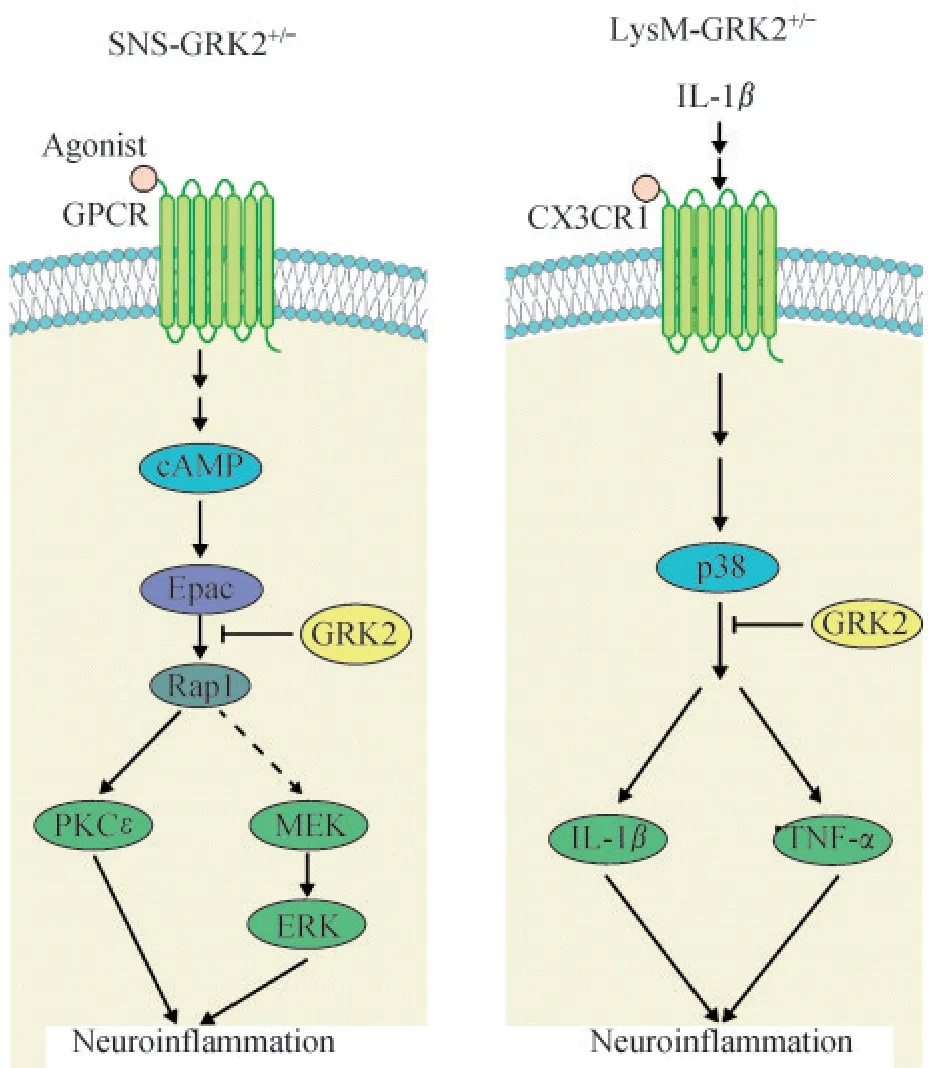
图2 GRK2 参与炎症痛的机制Fig 2 The mechanism of GRK2 involved in inflammatory pain
神经元GRK2 在炎症痛中的作用 研究发现,在足底注射角叉菜胶诱导的炎症痛模型中,与对照组相比,小直径初级感觉神经元内GRK2 水平降低约35%[22]。特异性敲减Nav1.8+外周感觉神经元的GRK2(SNS-GRK2+/-小鼠),角叉菜胶引起的热痛觉过敏可以延长至8 天,而野生型(wild type,WT)小鼠的热痛觉过敏仅持续2~4 天左右[23]。在SNSGRK2+/-小鼠中,EPI 诱导的机械痛和热痛觉过敏可延长至21 天左右,而在WT 小鼠中仅持续3~4天[24]。上述结果提示,初级感觉神经元GRK2 水平降低可能是急性炎症痛慢性化的核心机制之一。
研究表明,炎症痛时背根神经节(dorsal root ganglia,DRG)神经元 GRK2 水平降低可以增强Epac1-to-Rap1信号通路的活性,使cAMP信号传递到Epac1/Rap1。在弗氏完全佐剂(Freund’s complete adjuvant,FCA)诱导的小鼠炎症痛模型中,CFA 注射5 天后IB4+DRG 神经元内GRK2 水平明显降低,而DRG 内环磷酸腺苷直接激活的交换蛋白1(exchange protein directly activated by cAMP 1,Epac1)水平升高。与对照组相比,足底注射HSVGRK2 使得 DRG 神经元内过表达 GRK2 后 CFA 组小鼠炎症痛明显减轻;鞘内注射Epac1 反义寡核苷酸(antisense oligodeoxynucleotide,ASODN)下 调Epac1 也可以减轻 CFA 诱导的炎症痛[25]。其他研究[26]也显示,CFA 诱导的炎症痛小鼠脊髓内 GRK2降低,GRK2/Epac1 比值降低,脊髓神经元内提高GRK2 表达可显著减轻CFA 诱导的炎症痛。GRK2可以结合Epac1,通过磷酸化Epac1 的Ser-108 位点抑制Epac1 向质膜的易位,从而减少Ras 样小GTP酶 1(Ras-like GTPases 1,Rap1)的激活;小鼠发生炎症痛时 DRG 内 GRK2 降低,Epac1 靶向 Rap1 的活动增加。因此,GRK2 在炎症痛中的作用与Epac1 紧密相关。Epac1 下游的蛋白激酶Cε(protein kinase Cε,PKCε)和 MEK/ERK 依赖的信号通路在急性炎症痛转为慢性中发挥重要作用。在EPI/PGE2 诱导的小鼠炎症痛模型中,足底注射蛋白激酶A(protein kinase A,PKA)抑制剂 H-89 明显减少 WT 小鼠的炎症痛,对 SNS-GRK2+/-小鼠无影响;PKCε 抑制剂TAT-PKCεv1-2明显减少 SNS-GRK2+/-小鼠的炎症痛,对 WT 小鼠无影响;MEK 抑制剂 U0126 可以部分缓解WT 小鼠的急性期炎症痛,明显减少SNSGRK2+/-小鼠的炎症痛[22,24]。由此推断,感觉神经元GRK2 表达降低可将EPI/PGE2 诱导的信号从PKA转向PKCε 和MEK/ERK 依赖的信号通路,从而使急性炎症痛转为慢性[22,24]。有研究表明,P2X3 作为PKCε 下游在炎症痛时表达水平升高,P2X3 受体敏化导致ATP 电流产生增加,参与炎症痛的调节[27]。另外,Epac1 激活也可以增加机械敏感通道Piezo2介导的机械敏感电流,鞘内注射Piezo2 ASODN 下调DRG 内Piezo2 水平可以抑制CFA 诱导的机械性痛觉过敏,而过表达GRK2 可以抑制Epac1 介导的Piezo2 敏化[26],从而抑制炎症痛转为慢性。
此外,体外研究表明,神经元GRK2 减少可以增强 CCL3 诱导的 TRPV1 敏化,过表达 GRK2 则相反;在GRK2+/-鼠体内瞬时受体电位香草酸受体1(transient receptor potential vanilloid 1,TRPV1)参与介导CCL3 诱导的急性炎症痛,足底注射TRPV1拮抗剂预处理可以完全预防CCL3 诱导的急性炎症痛[23]。
小胶质细胞GRK2 在炎症痛中的作用 已有许多证据表明,小胶质细胞在急性痛转为慢性痛中起重要作用[28-29]。例如在大鼠神经病理性疼痛模型中,损伤同侧脊髓小胶质细胞激活,鞘内注射小胶质细胞抑制剂米诺环素可以预防慢性痛[30]。近年来的研究显示,与对照组相比,小鼠足底注射高剂量角叉菜胶(2%,20 μL)后脊髓小胶质细胞/巨噬细胞 GRK2 减 少约 40%[20]。在 LysM-GRK2+/-(小胶质细胞/巨噬细胞内特异性敲减GRK2)小鼠中,足底注射小剂量角叉菜胶(1%,5 μL)诱导的热痛觉过敏持续至少20 天,而在野生型鼠中2~4 天内就可消退[23];在角叉菜胶、CCL3、IL-1β 诱导的小鼠炎症痛模型中,GRK2+/-小鼠和LysM-GRK2+/-小鼠炎症痛持续时间基本相同,提示小胶质细胞/巨噬细胞GRK2 在调节这些类型炎症痛的持续时间上可能起决定作用[23]。与野生型小鼠相比,GRK2+/-小鼠足底注射角叉菜胶后第2 天引起的腰髓小胶质细胞/巨噬细胞激活更为显著[23]。鞘内注射小胶质/巨噬细胞抑制剂米诺环素进行预处理,虽然并不影响野生型小鼠和GRK2+/-小鼠足底注射角叉菜胶引起的急性炎症痛(<12 h),但可以显著抑制GRK2+/-小鼠足底注射角叉菜胶形成的慢性痛觉过敏;而在角叉菜胶注射后第7 天鞘内注射米诺环素可以显著缓解GRK2+/-小鼠的热痛觉过敏[23]。以上结果均表明,脊髓内小胶质细胞/巨噬细胞GRK2 的减少,参与了角叉菜胶引起炎症痛由急性期到慢性期的转变。
进一步研究表明,小胶质细胞/巨噬细胞GRK2可 能 通 过 CX3CR1、p38 以及 IL-1β 信 号 参 与 维 持IL-1β 引起的痛觉过敏和小胶质细胞/巨噬细胞的激活[20]。GRK2+/-小鼠足底注射 IL-1β 后第 2 天,腰髓CX3C 趋 化 因 子 受 体 1(chemokine CX3C motif receptor 1,CX3CR1)和 IL-1β mRNA 水平升高,鞘内注射抗CX3CR1 中和抗体或IL-1 拮抗剂IL-1ra可以缓解慢性痛[20]。小胶质细胞CX3CR1 激活后可以激活p38 并导致促炎性细胞因子如IL-1β 产生增多。体外实验证据表明GRK2 可以直接与p38 结合并使p38 的Thr-123 位点磷酸化,从而阻止上游激酶以及下游靶点的结合[31]。IL-1β 注射后第 2 天,鞘内注射p38 抑制剂SB239063 可以逆转LysMGRK2+/-小鼠的热痛觉过敏[20]。
研究表明,GRK2 对于炎症痛的调节与小胶质细胞极化分型有关。在LysM-GRK2+/-小鼠IL-1β 诱导的炎症痛模型中,急性到慢性炎症痛的转变与脊髓中M1 促炎型小胶质细胞/巨噬细胞持续性活化相关[32]。与 WT 小鼠相比,LysM-GRK2+/-小鼠足底注射IL-1β 后,脊髓小胶质细胞/巨噬细胞促炎细胞因子 IL-1β、TNF-α 显著增多、抗炎细胞因子 IL-10显著减少[32-33]。而进一步的研究表明,小胶质细胞的极化分型受到miR-124 的调节。与WT 小鼠相比,足底注射 IL-1β 后 24 h,LysM-GRK2+/-小鼠腰髓小胶质细胞miR-124 表达水平显著降低,鞘内注射miR-124 可以使M1 和M2 标志物表达恢复正常,并抑制IL-1β 诱导的持续性痛觉过敏。此外鞘内注射miR-124 也可以逆转高剂量角叉菜胶诱导的慢性痛[33]。
星形胶质细胞GRK2 在炎症痛中的作用 作为神经系统数量最多的一类细胞,近年来星形胶质细胞在疼痛中的作用被大量报道。研究发现,与野生型对照小鼠相比,尽管GFAP-GRK2+/-小鼠星形胶质细胞内 GRK2 水平降低约 60%[23],然而星形胶质细胞GRK2 的改变并不影响足底注射IL-1β、角叉菜胶、CCL3 诱导的热痛觉过敏[20,23]。并且,和对照组相比,GFAP-GRK2+/-小鼠中EPI 诱导的机械性痛觉过敏也没有显著差异[24]。星形胶质细胞中GRK2 是否参与其他疼痛模型还有待进一步研究。
GRK2 与镇痛已有研究表明,过表达GRK2可以防止PGE2 诱导的小鼠炎症痛的延长[25]。Epac特异性抑制剂ESI-09 可以抑制CFA 诱导的机械性痛觉过敏而不影响正常痛阈[26]。在持续的炎症痛期间,增加GRK2 或减少Epac1 可以抑制CFA 诱导的慢性痛觉过敏,提示靶向GRK2/Epac1 使GRK2/Epac1 保持稳态可以预防或治疗慢性痛[25]。我们前期的研究也表明,在CFA 诱导的小鼠炎症痛模型中,电针能够通过提高脊髓背角GRK2/Epac1 的比例,使脊髓背角miR-124 表达上调,抑制M1 型小胶质细胞的活化,促进M2 型小胶质细胞的活化,从而缓解炎症痛;降低GRK2 表达可以抑制电针的镇痛以及对miR-124 和小胶质细胞的正向调节作用[34]。
除炎症痛模型外,也有研究表明,在癌痛模型和神经病理性疼痛模型中脊髓GRK2 表达水平降低[12,35-36]。 鞘 内 或 腹 腔 注 射 大 麻 素 2 受 体 激 动 剂JWH-015 可恢复脊髓GRK2 表达水平,并减轻Walker 256 乳腺癌细胞接种诱导的大鼠胫骨癌痛[35]。鞘内注射特异性抑制剂下调miR-15a 和miR-16 可显著增加CCI 大鼠脊髓GRK2 的表达,明显减轻CCI 大鼠的机械性痛觉超敏和热痛觉过敏[36]。以上研究结果均提示升高脊髓GRK2 可能可以缓解癌痛和CCI 神经病理性疼痛,GRK2 可以成为疼痛治疗的一个潜在新靶点。然而,目前能够恢复脊髓GRK2 的治疗方法只有如大麻素受体激动剂[35]、miR 拮抗剂[36]、针刺疗法[34]等少量报道,更多的治疗药物或方法仍有待进一步研究开发。此外,也有研究显示GRK2 参与神经元阿片受体的脱敏过程[37-38]。在生理状态下,GRK2 可能长期与 δ 阿片受体结合,下调外周感觉神经元δ 阿片受体的抗伤害感受性,导致正常状态下外周δ 阿片受体功能不全。敲低 GRK2 使 δ 阿片受体激动剂 DPDPE 对 δ 阿片受体的激活作用增强,恢复δ 阿片受体的抗伤害感受性[38]。但在疼痛状态下过表达GRK2 是否影响阿片受体激动剂的镇痛效果仍需要进一步研究。
结语目前对GRK2 在炎症痛中的基础和临床研究仍然较少,且基础研究多集中在外周感觉神经元和小胶质细胞中,而针对中枢神经系统神经元和星形胶质细胞中GRK2 的研究相对较少。此外,现阶段的研究表明GRK2 主要在急性炎症痛转变为慢性炎症痛中发挥作用,但相关机制的研究仍然较少,有待进一步探索。有关GRK2 的研究可为炎症痛发病机制的分析和相关新药的研发提供新思路。随着炎症痛机制研究的不断深入,必然为炎症痛的治疗提供新的靶点,进而开发更多、更有效的药物,提高炎症痛患者的生活质量。
作者贡献声明李晓晨 综述构思及撰写,图表绘制。陈愈,王彦青 综述修订。毛应启梁 综述构思及修订。
利益冲突声明所有作者均声明不存在利益冲突。
猜你喜欢
世界中医药(2021年14期)2021-09-13 02:41:50
中老年保健(2021年7期)2021-08-22 07:40:34
神经损伤与功能重建(2020年11期)2020-12-01 05:01:54
家庭医学(下半月)(2020年4期)2020-05-30 12:42:40
家庭医学(下半月)(2020年4期)2020-05-30 12:42:40
湖南中医药大学学报(2016年1期)2016-12-01 04:08:21
好孩子画报(2016年4期)2016-11-19 08:41:24
磁共振成像(2015年1期)2015-12-23 08:52:21
实验动物与比较医学(2014年4期)2014-02-28 14:52:58
河北医科大学学报(2011年1期)2011-03-25 10:15:37
