
高效液相色谱法测定利巴韦林滴眼液中有关物质的含量
2021-10-09 08:15冯文刘峰谢华四川省药品检验研究院成都611731
中南药学 2021年9期
冯文,刘峰,谢华(四川省药品检验研究院,成都 611731)
利巴韦林(ribavirin)又名病毒唑、三氮唑核苷等,化学名为1-β-D-呋喃核糖基-1H-1,2,4-三唑-3-羧酰胺(结构式见图1),是广谱强效的抗逆转录病毒药物[1-2],属合成核苷类药,对多种DNA 和RNA 病毒有抑制作用,具有作用位点多、不易产生耐药性、疗效高、毒性低和不良反应少等特点,现已用于多种病毒的治疗。
有关物质的考察贯穿于整个药品研究,直接关系到制剂质量的可控性与安全性。利巴韦林滴眼液的有关物质测定方法未见报道,国内外药典虽收载其原料药的有关物质检测方法[3-6],但由于滴眼液中抑菌剂等特殊辅料[7](如羟苯乙酯)与利巴韦林极性差异较大,利巴韦林原料药的有关物质测定方法无法及时将其洗脱,连续进样多针后残留物将干扰后续样品的测定。因此,本文建立了一种测定利巴韦林滴眼液中主要降解杂质A(结构式见图1)及其他有关物质的方法,以消除滴眼液中特殊辅料的干扰,为其质量提供了可靠的检测依据,也为本品研发、生产和质量提升提供分析方法。
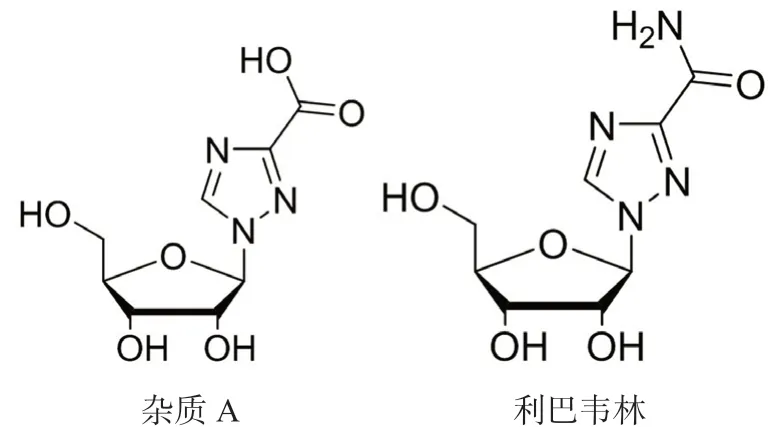
图1 利巴韦林杂质A 与利巴韦林化学结构式Fig 1 Chemical structure formula of impurity A and ribavirin
1 仪器与试药
高效液相色谱仪Agilent 1100(安捷伦科技有限公司);高效液相色谱仪岛津LC20A(岛津实验器材有限公司);电子天平(赛多利斯Sartorius CPA255D,十万分之一)。
利巴韦林滴眼液[A 企业3 批次;B 企业15批次;C 企业2 批次;D 企业2 批次;E 企业6 批次;F 企业15 批次;G 企业14 批次;H 企业5 批次;I 企业3 批次;J 企业14 批次;K 企业17 批次(其中批号为20010401 样品用于方法学验证)]。利巴韦林对照品(中国食品药品检定研究院,批号:140629-201703,含量:99.60%),利巴韦林杂质A(TLC,批号:3793-019A1,含量:100%),空白辅料由各企业提供。
2 方法与结果
2.1 溶液的制备
2.1.1 供试品溶液的制备 取本品适量,用水稀释制成每1 mL 中约含利巴韦林0.5 mg 的溶液,作为供试品溶液。
2.1.2 对照品储备液的制备 分别精密称取利巴韦林对照品与杂质A 对照品各约10 mg,置100 mL 量瓶中,加水溶解并稀释至刻度,摇匀,即得利巴韦林对照品储备液与杂质A 对照品储备液。
2.1.3 对照品溶液的制备 精密量取利巴韦林对照品储备液与杂质A 对照品储备液各2.5 mL,置同一100 mL 量瓶中,用水稀释至刻度,摇匀,作为对照品溶液。
2.1.4 对照溶液的制备 精密量取供试品溶液1 mL,置100 mL 量瓶中,用水稀释至刻度,摇匀,作为对照溶液。
2.1.5 系统适用性溶液 取供试品溶液5 mL,加入1 mol·L-1的氢氧化钠溶液5 mL,静置30 min,用5 mL 1 mol·L-1的盐酸溶液中和,以产生杂质A 溶液作为系统适用性溶液。
2.1.6 空白辅料溶液配制 由于各企业处方不同,空白辅料溶液是11 家企业所用所有辅料的混合溶液。即精密称取氯化钠0.9 g、硼酸0.95 g、硼砂50 mg、羟苯乙酯40 mg、苯扎溴铵10 mg、甘油1.0 g 置100 mL 量瓶中,加水溶解并稀释至刻度,摇匀;精密量取1 mL 置10 mL 量瓶中,用水稀释至刻度,摇匀,即得。
2.2 色谱条件与系统适用性
以十八烷基硅烷键合硅胶为填料的色谱柱Waters Atlantis T3(250 mm×4.6 mm,5 μm);以0.1%硫酸钠水溶液(取无水硫酸钠1.0 g,加水950 mL 溶解,加2 mL5%磷酸,用5%磷酸调节pH 至2.8,用水稀释至1000 mL,即得)为流动相A,以乙腈-流动相A(5∶95)为流动相B,以20%乙腈为流动相C,以乙腈为流动相D;流速为1.0 mL·min-1;按表1进行梯度洗脱;柱温为30℃;检测波长为220 nm;进样体积为20 μL。系统适用性溶液中,杂质A(相对保留时间约0.78)与利巴韦林分离度应不低于4.0。

表1 梯度洗脱程序Tab 1 Gradient elution
2.3 专属性考察
精密量取空白辅料溶液、系统适用性溶液及供试品溶液各20 μL 进样,记录色谱图(见图2)。空白辅料对本品有关物质的测定无干扰,各杂质与主成分分离良好,方法专属性良好。
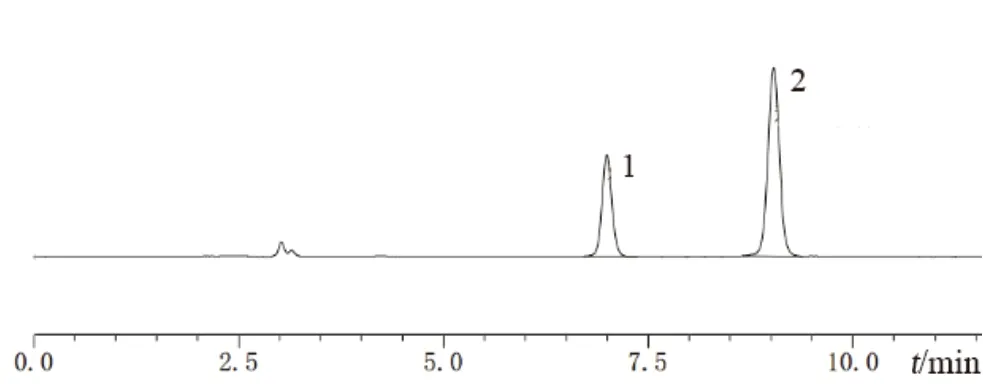
图2 系统适用性溶液图谱Fig 2 HPLC chromatogram of system suitability solution
2.4 强制降解试验
为考察在所选择的色谱条件下能否检出利巴韦林滴眼液中可能产生的降解产物,分别用高温、酸、碱、氧化和光照等剧烈条件对本品进行破坏,试验如下:
① 未破坏溶液:精密量取本品5 mL,置10 mL 量瓶中,加水稀释至刻度,摇匀。
② 酸破坏:精密量取本品5 mL,置10 mL量瓶中,加1 mol·L-1盐酸溶液2 mL,静置120 min,用等浓度氢氧化钠溶液中和,加水稀释至刻度作为酸破坏溶液;同法配制酸空白溶液及酸破坏辅料溶液。
③ 碱破坏:精密量取本品5 mL,置10 mL 量瓶中,加1 mol·L-1氢氧化钠溶液2 mL,静置60 min,用等浓度盐酸溶液中和,加水稀释至刻度作为碱破坏溶液;同法配制碱空白溶液及辅料溶液。
④ 高温破坏:精密量取本品5 mL,置10 mL量瓶中,加水稀释至刻度,摇匀,密封,于105℃加热8 h 作为高温破坏溶液;同法配制高温辅料溶液。
⑤ 光照破坏:取本品适量,5000 Lx 光照射5 d,取破坏后样品5 mL,置10 mL 量瓶中,加水稀释至刻度,摇匀;同法配制光照辅料溶液。
⑥ 氧化破坏:精密量取本品5 mL,置10 mL 量瓶中,加入30%过氧化氢溶液2.0 mL,静置60 min,用水稀释至刻度,摇匀,作为氧化破坏溶液;同法配制氧化空白溶液及辅料溶液。
取上述溶液各20 μL,分别注入高效液相色谱仪,结果见图3。由强制降解试验结果可知,本品在光照条件下相对比较稳定;在酸、碱和高温条件下主要破坏产生杂质A,其中在碱性条件下破坏产生的杂质A 最多;在氧化条件下破坏出杂质峰个数最多,保留时间约为5.5 min 和17 min 的杂质有明显增加。破坏试验中各杂质峰与利巴韦林峰分离良好,无其他杂质峰干扰已知杂质的测定,能有效检出杂质,表明该色谱方法专属性良好。
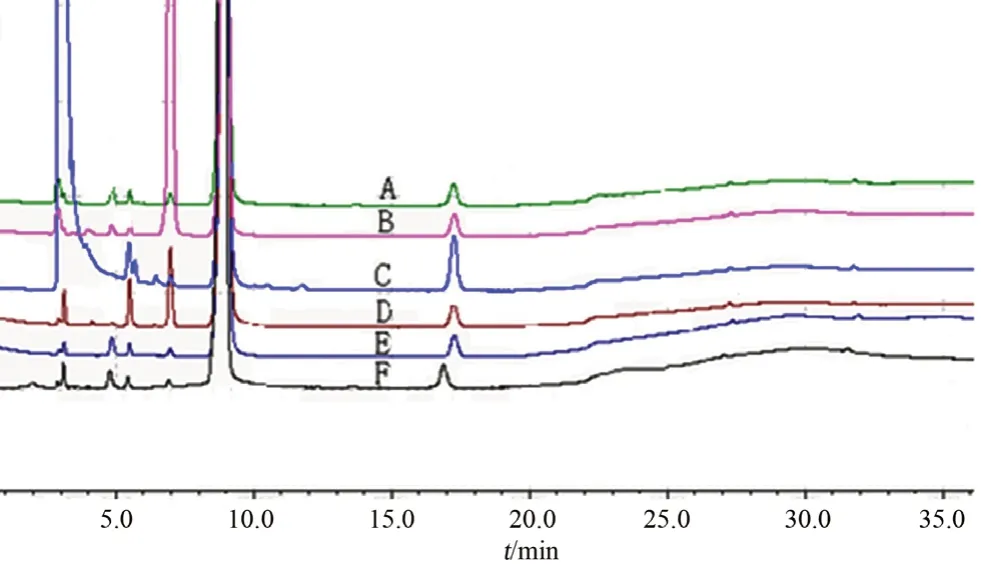
图3 强制降解试验色谱图Fig 3 HPLC chromatogram of forced degradation test
2.5 线性试验
精密量取“2.1.2”项下对照品储备液适量,用水稀释制成含利巴韦林和杂质A 的质量浓度分别约为0.25、0.5、2.5、5.0、10.0、15.0 μg·mL-1的系列浓度的对照品溶液。精密量取系列浓度的对照品溶液各20 μL,分别注入高效液相色谱仪,记录色谱图。以质量浓度作为横坐标,峰面积作为纵坐标,绘制标准曲线,见表2。

表2 利巴韦林和杂质A 的回归方程、范围、检测限和定量限Tab 2 Linearity,regression equation,LOQ and LOD of ribavirin and impurity A
2.6 检测限、定量限
将线性溶液(15 μg·mL-1)逐步稀释后进样测定,以色谱图中信噪比(S/N)为3∶1 确定检测限,S/N为10∶1 确定定量限,结果见表2。
2.7 进样精密度
精密吸取对照品溶液与对照溶液各20 μL,按“2.2”项下色谱条件测定6 次,记录色谱图,考察进样精密度,结果利巴韦林与杂质A 连续6针峰面积RSD分别为0.3%和0.4%,表明本法进样精密度良好。
2.8 稳定性
精密吸取对照品溶液、对照溶液与供试品溶液各20 μL,按“2.2”项下色谱条件分别在常温下于0、2、4、6、10、24 h 进样,记录色谱图,结果主成分及杂质A 峰面积RSD均小于2%,也无其他杂质产生,表明各溶液在24 h 内稳定。
2.9 准确度
按照各企业提供的处方及辅料对试药进行混合,得到多组分多厂家共用空白辅料,添加相应的利巴韦林对照品(杂质均未检出),作为阴性样品,称取阴性样品适量(约相当于利巴韦林5 mg),共9 份,分别置10 mL 量瓶中,再分别精密加入对照品储备液0.8、1.0、1.2 mL,各3份,加流动相适量使溶解并稀释至刻度,摇匀,滤过,取续滤液作为准确度试验80%、100%、120%的供试品溶液,按照拟订的色谱条件检测。结果杂质A 的回收率在94.6%~100.3%,RSD均<2.0%,表明该方法准确度较高。
2.10 重复性
精密量取本品适量,用水定量稀释制成每1 mL 中约含利巴韦林0.5 mg 的溶液,作为供试品溶液,平行制备6 份,按“2.2”项下方法测定,已知杂质A 按外标法以峰面积计算含量,未知杂质按自身对照法以峰面积计算含量,结果显示,杂质A的RSD为4.1%,最大未知单杂RSD为3.3%,总杂RSD为4.1%,表明方法重复性良好。
2.11 杂质校正因子
在两台液相色谱仪(Agilent 1100 高效液相色谱仪和岛津LC20A 高效液相色谱仪)上,考察3 根色谱柱[型号分别为:Waters Atlantis T3(250 mm×4.6 mm,5 μm)、Waters Symmetry Shield C18(4.6 mm×150 mm 5 μm)、YMC-Triart C18(4.6 mm×150 mm,3 μm)],分别进样“2.5”项下线性溶液,以各成分进样量对峰面积进行回归,用利巴韦林斜率除以杂质A 斜率测得杂质A 相对校正因子的平均值为2.0,为杂质A 的准确定量提供了经济便捷的计算方式。
2.12 耐用性
为考察本方法对条件发生微小变化的耐受程度,进行耐用性试验,改变因素包括流动相比例±2%、流速变化±10%、柱温变化±10%。以含有3 种已知杂质A 的样品(D 企业,批号20010401)为耐用性试验溶液,考察条件改变对测定结果的影响。经测定,在上述各条件下杂质A 与主成分之间分离度良好,测得杂质A 的RSD分别为3.4%,最大未知单杂RSD为4.5%,总杂RSD为3.3%,表明方法耐用性良好。
2.13 样品测定
按“2.3”项下方法测定11 个不同生产企业共96 批利巴韦林滴眼液中的有关物质,结果见表3。
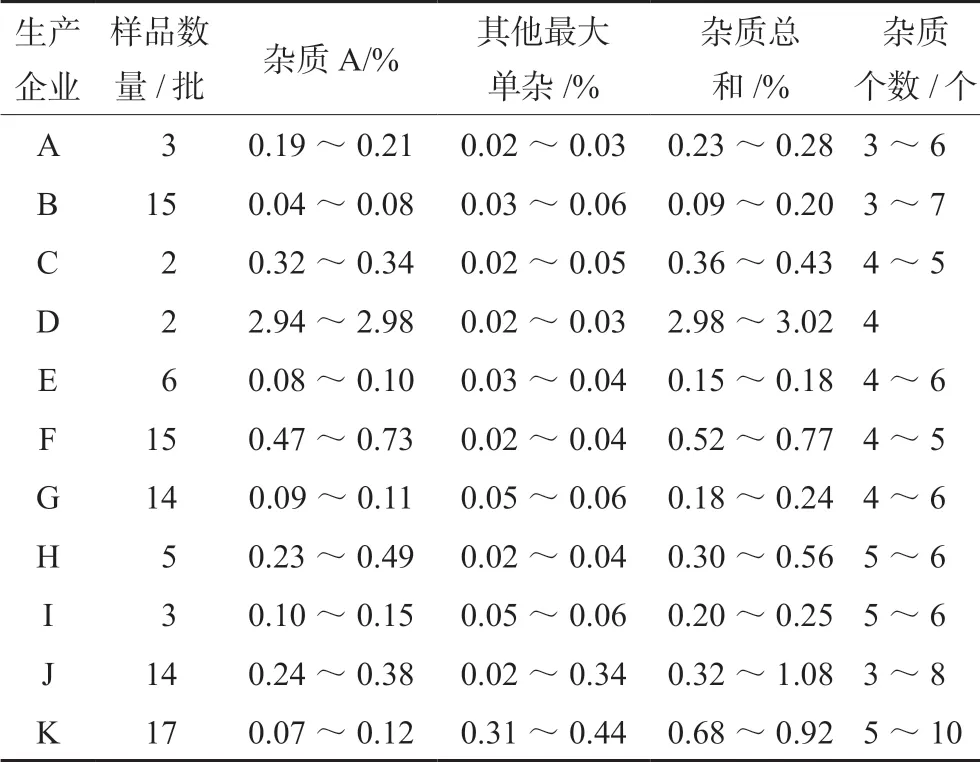
表3 利巴韦林滴眼液有关物质检查结果Tab 3 Determination of related compounds in ribavirin eyes drops
2.14 限度
参照《中国药典》2020年版利巴韦林原料及其注射液的限度,同时考虑本品的临床使用情况,拟定有关物质限度为:供试品溶液色谱图中如有杂质峰,除辅料峰外,杂质A 按校正后的峰面积计算(乘以校正因子2.0)不得大于对照溶液主峰面积的0.5 倍(0.5%);其他单个杂质峰面积不得大于对照溶液主峰面积的0.5 倍;各杂质校正后峰面积的和不得大于对照溶液主峰面积(1.0%)。
2.15 未知杂质的定性研究
K 企业17 批样品均合格,其中最大未知单杂均为相对保留时间约1.9 的杂质,检出量为利巴韦林标示量的0.32%~0.44%,其他10 家生产企业样品中未检出该杂质。采用2D-HPLC-DADESI-Q-TOF 技术[一维液相条件同本文建立的色谱条件;二维液相条件为:采用中心切割,色谱柱为Agilent EclipsePlus C18(3.0 mm×150 mm,1.8 μm);流动相为以0.1%甲酸水溶液为流动相A,以甲醇为流动相B,梯度洗脱(0~17.6 min,95%A;17.6~23 min,95%→5%A;23~35 min,5%A),流速为0.3 mL·min-1,检测波长为220 nm;质谱条件为:采用ESI 源,质谱扫描范围为100~1000m/z,负离子模式,Gas Temp:325℃,Drying Gas:8 L·min-1,VCap:3500 V,Expt:1000 V]。推测降解反应机制,将K 企业样品(批号:20010401)105℃加热8 h 进行分析,结果见图4。

图4 未知杂质(相对保留时间约1.9)的一维液相色谱图(A)和质谱图(B)Fig 4 One-dimensional liquid chromatogram(A)and MS1(B)of unknown impurity(relative retention time about 1.9)
根据质谱结果,相对保留时间约为1.9 的杂质m/z为200.9832[M-H]-,推测其为硫柳汞钠与氯化钠配伍所产生[8-10],反应机制为:硫柳汞钠在氯化钠存在的条件下,可分解为硫代水杨酸和氯化乙基汞,硫代水杨酸不能稳定存在,经氧化生成较为稳定的2,2'-二硫代二苯甲酸。2,2'-二硫代二苯甲酸在乙基汞的作用下,部分分解转化为硫柳汞钠,部分经三步氧化,最终转化为2-磺基苯甲酸(见图5)。
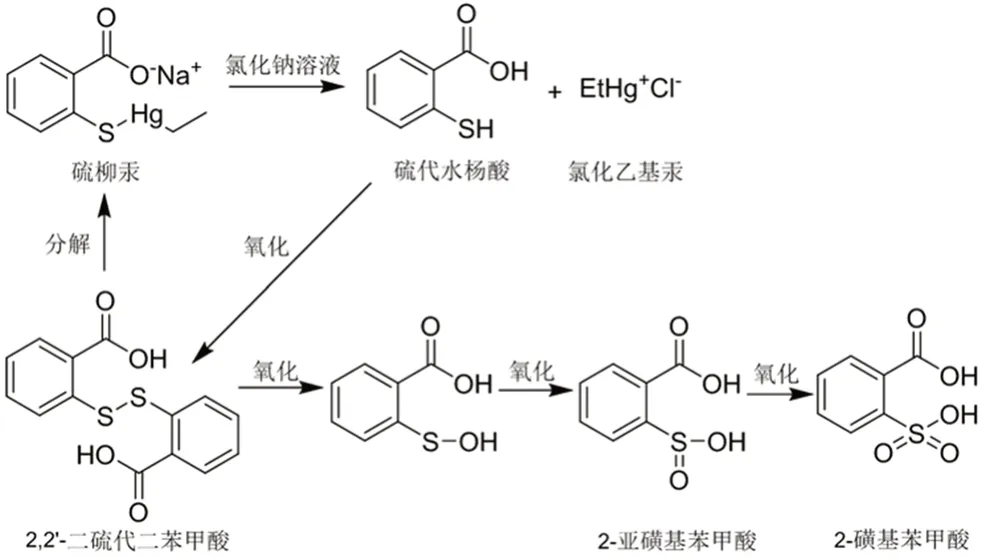
图5 2-磺基苯甲酸生成机制图Fig 5 Formation mechanism of 2-sulfobenzoic acid
3 讨论
3.1 色谱条件筛选
采用利巴韦林的碱破坏溶液,分别参照ChP2020、BP2020 中利巴韦林有关物质检查法进行试验。ChP2020 采用氢型阳离子交换柱,在该色谱条件下主峰出峰较快,峰形较差,与相邻杂质峰分离度不佳,且由于该类色谱柱与有机溶剂不能耐受,无法洗脱滴眼液中抑菌剂等特殊辅料(如羟苯乙酯),连续进样多针后对后续样品测定造成干扰。故选择在条件相对较优的BP2020 基础上对色谱柱和洗脱程序等进一步优化。分别考察以下色谱柱(1)Waters Xbridge C18(4.6 mm×250 mm,5 μm);(2)J&K C18(4.6 mm×250 mm,5 μm);(3)Waters Atlantis T3(4.6 mm×250 mm,5 μm)。发现采用色谱柱(1)和(2)时,利巴韦林由于其极性强,保留时间较短,与碱破坏产生的杂质A 分离度不能达到4.0。采用色谱柱(3)时,利巴韦林峰保留时间约为9 min,杂质A 与主成分峰间分离度约为8.2,各杂质间分离度均良好。在此基础上调节梯度洗脱程序,由于利巴韦林与滴眼液中特殊辅料如羟苯乙酯极性差异较大,因此本法增加乙腈梯度洗脱的程序。
3.2 不合格样品原因分析
以拟订限度判断,本次抽取的96 批样品有关物质合格率为81.2%,其中,D 企业2 批样品均不合格,均为杂质A 超出限度,其检出量为2.94%~2.98%;F 企业15 批样品中14 批不合格,均为杂质A 超出限度,其检出量为0.53%~0.73%;J 企业14 批样品中2 批不合格,均为总杂超出限度,其检出量为1.06%~1.08%。
不合格样品主要由杂质A 含量超出限度(0.5%)导致,通过强制降解试验及影响因素试验可知,杂质A 为利巴韦林的主要降解杂质,高温、氧化、碱性条件均会导致其大量产生。分析企业处方和工艺流程,如D 企业处方中含有氢氧化钠(约3.75 μg·mL-1),生产工艺中采用沸水溶解原辅料,可能出现高温条件下局部碱性过强,导致利巴韦林降解产生杂质A。因此,建议企业在生产工艺过程中关注原辅料的溶解温度,避免用煮沸的方式使其溶解。同时,在使用氢氧化钠等碱性pH 调节剂时注意其加入方式,避免局部碱性过浓造成利巴韦林的降解。
3.3 J 企业样品生产时间与杂质量相关性分析
J 企业的14 批样品测定结果显示相对保留时间约为0.62 的杂质的检出量为0.02%~0.34%,查阅其批号,发现含量为0.02%的样品批号为200301(生产日期为2020年3月),含量为0.34%的样品批号为190703(生产日期为2019年7月),同时期生产的样品有关物质含量相当(见图6),提示该企业杂质含量随贮存时间延长增长趋势较为明显,存在质量隐患。
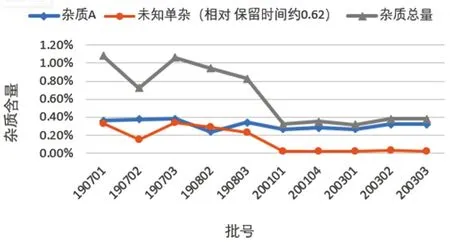
图6 J 企业质随批号变化趋势图Fig 6 Impurity change trend with batch number of J company
3.4 抑菌剂使用合理性分析
采用有关物质检查方法,K 企业的17 批样品中,2-磺基苯甲酸检出量为利巴韦林标示量的0.32%~0.44%。按硫柳汞钠标示量计算,则约为16%~22%,硫柳汞钠大量降解,可能样品中存在一定量的硫柳汞钠分解产生的氯化乙基汞,氯化乙基汞为剧毒物质[11],其中人经口吸收,最小致死剂量为30 mg·kg-1,1 mg·L-1可致人Hela 细胞突变,同时还具有一定的生殖毒性。氯化乙基汞可经口、呼吸道、皮肤和黏膜吸收,大剂量接触可造成急性中毒,低剂量长期接触可造成慢性中毒[12],对人的神经、心脏、皮肤等系统造成严重损害,存在较大安全隐患。因此建议硫柳汞钠从处方中去除,且处方中不应与氯化钠配伍使用。
猜你喜欢
人人健康(2021年14期)2021-08-06
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
家庭医学(2018年8期)2018-10-17
山东化工(2018年15期)2018-09-20
中国卫生标准管理(2015年16期)2016-01-20
中国继续医学教育(2015年6期)2016-01-07
首都食品与医药(2015年18期)2015-11-03
药学与临床研究(2015年4期)2015-06-05
药学研究(2012年2期)2012-10-25
