
单核巨噬细胞在心肌炎中的作用
2021-10-08 06:14:46路阳赵宁杜以梅
中国心血管杂志 2021年4期
路阳 赵宁 杜以梅
430022 武汉,华中科技大学同济医学院附属协和医院心内科
心肌炎是一种以心脏炎症为特征的心脏病,是扩张型心肌病(dilated cardiomyopathy,DCM)的主要病因之一。心肌炎最常由柯萨奇病毒B3(coxsackievirus B3,CVB3)、腺病毒和细菌等感染性因素引起,毒素、酒精、药物和自身免疫病也会诱发心肌炎[1-2]。以往研究表明,T细胞是心肌炎中发挥主导作用的免疫细胞,然而,单核巨噬细胞在心脏浸润的免疫细胞中占比最高[3],并发挥多种效应功能。在本综述中,我们总结了关于心脏单核细胞和巨噬细胞起源的最新研究,重点阐述了它们在心肌炎时数量和表型的动态变化,以及它们在抗病毒、炎症损伤、适应性免疫及组织修复中所起的作用,强调了以单核巨噬细胞为治疗靶点的临床应用价值。
1 心脏单核细胞和巨噬细胞的来源、表型和功能
单核细胞起源于骨髓中的单核细胞前体,成熟后进入血管和组织中,接受抗原后转移至淋巴结或在组织中分化为巨噬细胞。小鼠单核细胞由Ly6C从高到低的表达分为:Ly6Chigh、Ly6Cint和Ly6Clow3种单核细胞[4](表1)。Ly6Chigh单核细胞高表达Ly6C和CCR2,有较强的吞噬和促炎能力,被称为“炎性单核细胞”。Ly6Clow单核细胞低表达Ly6C,高表达CX3CR1,由Ly6Chigh单核细胞分化而来,参与巡逻、免疫监视和组织修复,被称为“巡逻单核细胞”。Ly6Cint单核细胞是Ly6Chigh单核细胞向Ly6Clow单核细胞转化的过渡阶段,具有一定的抗原提呈和促进血管生成能力。人类单核细胞根据CD14和CD16的表达也分为3个亚群[4](表1),CD14++CD16-经典型单核细胞功能类似于小鼠Ly6Chigh单核细胞,CD14+CD16++非经典型单核细胞类似于小鼠Ly6Clow单核细胞,而CD14++CD16+中间型单核细胞类似于小鼠Ly6Cint单核细胞。
过去,巨噬细胞通过其表面分子被分为M1型和M2型,M1型巨噬细胞促进炎症反应而M2型巨噬细胞参与抗炎和组织修复。然而,由于巨噬细胞功能的复杂性,这种M1/M2分类方法不够完善,因此近来有学者提出通过起源和表面标记来综合定义巨噬细胞亚群。心脏巨噬细胞有3种起源:原始的卵黄囊、胎儿肝脏单核细胞、骨髓单核细胞[5](表1)。前两者统称为胚胎来源的巨噬细胞,它们高表达CX3CR1,低表达CCR2,被称为CCR2-常驻巨噬细胞,是稳态时心脏巨噬细胞的主要成分,并通过原位增殖维持数量平衡。CCR2-巨噬细胞吞噬凋亡细胞,促进组织修复、血管生成和心脏电传导,抑制炎症细胞募集。而骨髓单核细胞来源的巨噬细胞高表达CCR2,在稳态心脏内含量较少,但在炎症时大量募集到心脏。CCR2+巨噬细胞高表达炎症基因,并通过NLPR3途径分泌白细胞介素(interleukin,IL)1β,分泌趋化因子促进炎症细胞的募集,造成炎症损伤和不良重构。
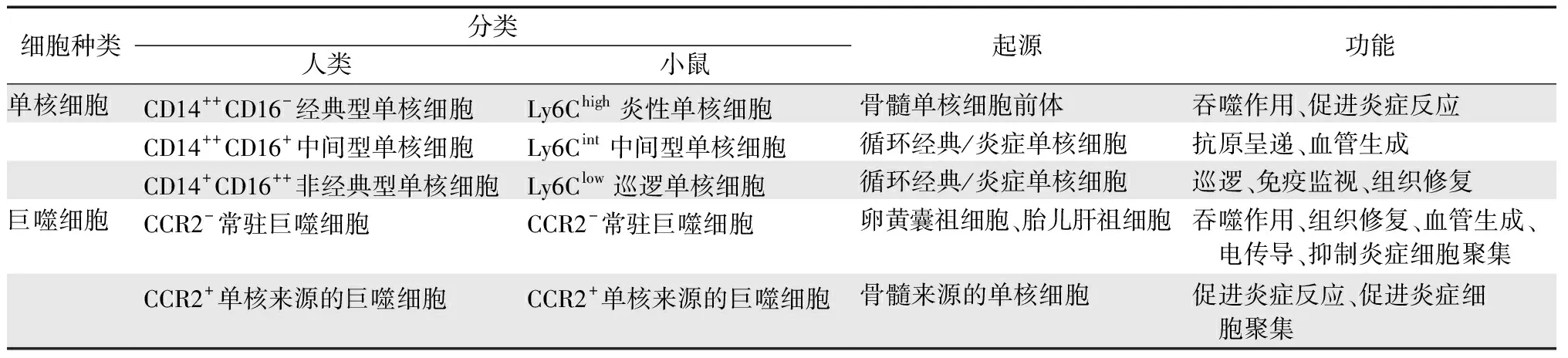
表1 心脏单核细胞和巨噬细胞的分类、起源和功能
2 单核巨噬细胞在心肌炎中的动态变化
大多数心肌炎研究集中在病毒性心肌炎(viral myocarditis,VMC)和实验性自身免疫性心肌炎(experimental autoimmune myocarditis,EAM)动物模型,因此我们主要总结这些模型中单核细胞和巨噬细胞的动态变化。在VMC和EAM的急性期,骨髓和脾脏中的Ly6Chigh单核细胞通过CCR2/CCL2等趋化因子轴浸润至心脏,在心脏增殖并分化为CCR2+MHC Ⅱ+巨噬细胞,其数量可达稳态时的8倍,占所有心脏浸润免疫细胞的60%以上,并发挥多种效应功能[3-6]。与之相反,在VMC小鼠急性期观察到胚胎来源的常驻巨噬细胞的减少,其数量在第7天约为稳态时的1/2[6],表明常驻巨噬细胞可能在VMC过程中发生凋亡,而它们在EAM模型中的变化尚无研究。在VMC和EAM慢性期,Ly6Chigh单核细胞凋亡或转变为Ly6Clow单核细胞,Ly6Clow单核细胞通过CX3CR1/CX3CL1轴募集到心脏,心脏CCR2+巨噬细胞数量和比例均下降,CCR2-巨噬细胞数量比例相对上升,在心脏中占主要地位,促进组织修复[6-7]。综上所述,单核巨噬细胞的动态变化大致为从急性期的以促炎性单核巨噬细胞为主过渡到慢性期的以修复性单核巨噬细胞为主的过程。然而,由于病毒基因组的持续存在和自身免疫的激活,导致巨噬细胞过度积累和转化障碍,造成心肌炎症损伤和纤维化加重,引起DCM。
3 单核巨噬细胞在心肌炎中的功能
3.1 清除病毒和坏死心肌
单核巨噬细胞在病毒和坏死心肌的清除中发挥重要作用(图1)。巨噬细胞表面Toll样受体(toll-like receptor,TLR)3是调控病毒感染的主要分子。研究表明,敲除心肌细胞上的TLR3并不影响它们对CVB3的易感性以及IFN-α/β基因的表达,因此心肌细胞上的TLR3可能不参与抗病毒反应[8]。相反,过继转输TLR3敲除的巨噬细胞的VMC小鼠相比转输正常巨噬细胞的小鼠,其心肌病变程度和血清肌钙蛋白I水平明显升高,表明巨噬细胞上TLR3参与了VMC抗病毒免疫[9]。因此,调控巨噬细胞TLR3的表达可有效改善病毒清除。
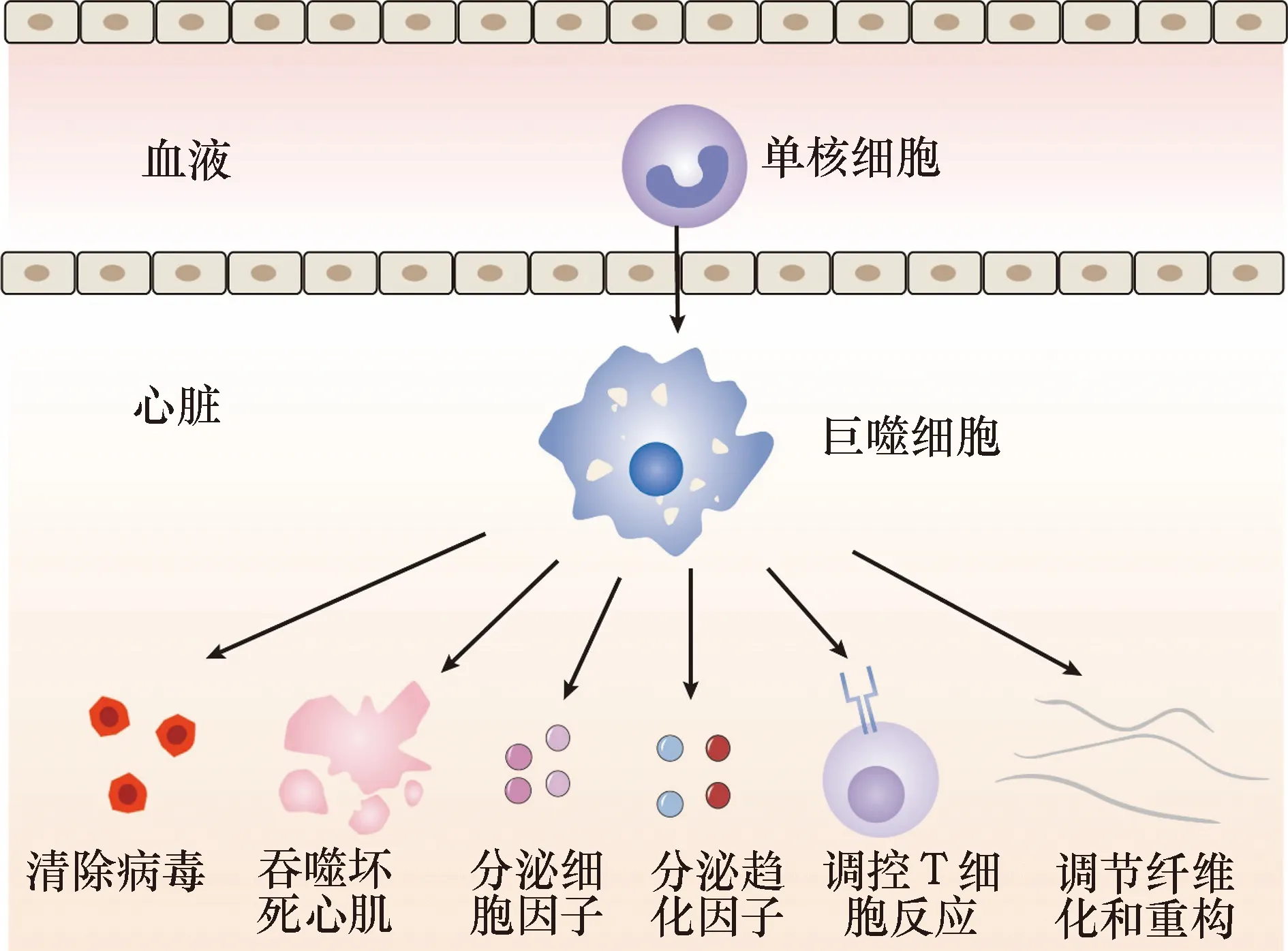
血液中的单核细胞进入心脏后分化为巨噬细胞,巨噬细胞发挥清除病毒、吞噬坏死心肌、分泌细胞因子和趋化因子、调控T细胞反应及调节纤维化和重构作用图1 单核巨噬细胞在心肌炎中的功能
除了清除病毒,巨噬细胞还通过其表面吞噬受体MerTK识别和清除坏死心肌,同时减少炎症因子分泌,这一过程称为胞葬作用。研究发现,心脏常驻CCR2-MHClow巨噬细胞表达更高水平的MerTK,它们在心肌损伤后识别和吞噬凋亡细胞,并分泌转化生长因子β(transforming growth factor-β,TGF-β)和IL-10抑制炎症反应,促进心脏修复,而骨髓来源的CCR2+单核巨噬细胞促进MerTK裂解,阻碍巨噬细胞的胞葬作用[10]。人体试验表明,与缺血性心肌病患者相比,心肌炎患者血液CD11b+髓系细胞中MerTK的表达明显降低,提示心肌炎时可能存在单核巨噬细胞吞噬功能障碍;小鼠实验证实,EAM小鼠心脏浸润的淋巴细胞分泌的IL-17刺激成纤维细胞分泌粒细胞-巨噬细胞集落刺激因子(granulocyte macrophage colony stimulating factor,GM-CSF),使巨噬细胞表面MerTK脱落,造成凋亡细胞清除和组织修复受阻[7]。因此,提高巨噬细胞表面MerTK的水平可作为促进坏死心肌清楚和组织修复的有效手段。
3.2 调节炎症和心肌损伤
单核巨噬细胞通过分泌细胞因子和趋化因子介导心脏炎症反应(图1)。在接受TLR配体或IFN-γ刺激后,巨噬细胞会分化为M1型,M1型巨噬细胞合成诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)及活性氧介导氧化应激,形成炎症小体并分泌TNF-α、IL-1β、IL-6、IL-8、IL-12和IL-18等细胞因子促进炎症反应;而M2型巨噬细胞被IL-4诱导分化,通过分泌IL-10和TGF-β等减轻炎症。同时,单核巨噬细胞还分泌多种集落刺激因子如M-CSF、GM-CSF,及趋化因子如CCL2、CCL5、CCL7、CXCL12、CX3CL1,促进自身或其他免疫细胞的增殖和浸润,引起炎症级联反应,造成心脏损伤[11]。
TLR可作为调控巨噬细胞功能的主要靶点。研究表明,人类心脏肌球蛋白及其肽段可直接激活外周血单核细胞上的TLR2,诱导细胞因子的分泌,引起炎症细胞的募集和激活[12]。小鼠实验显示,CVB3的衣壳蛋白可能通过激活TLR4使巨噬细胞形成炎症小体,促进IL-1β、IL-18的释放[13],而抑制TLR4可阻止上述因子的分泌,减轻病毒复制和心肌炎症[14]。
另一方面,调控M1/M2巨噬细胞平衡可作为治疗心肌炎的有效手段。研究发现,M1/M2巨噬细胞比例的不同是造成心肌炎损伤的性别差异的主要原因,雄性小鼠心肌炎严重程度高于雌性小鼠,雄性小鼠心脏浸润的巨噬细胞高表达M1型标志物iNOS、TNF-β、IL-12,而雌性小鼠巨噬细胞高表达M2型标志物Arg-1、IL-10、MMR[15]。研究者通过体外沉默巨噬细胞高迁移率族蛋白1(high mobility group protein box 1,HMGB1)的表达,抑制NF-κB、p38和Erk1/2通路的激活,减少巨噬细胞炎症因子的分泌,在EAM小鼠中沉默HMGB1后,心脏浸润的M1样巨噬细胞数量减少,并保护了心脏功能[16]。大量动物研究证实,通过调控各种不同的分子靶点或非编码RNA如AngⅡ、lncRNA AK085865,均可通过诱导M1型巨噬细胞向M2型巨噬细胞转化,抑制VMC或EAM的炎症损伤[17-18]。
靶向单核巨噬细胞增殖和浸润也可调控心脏炎症。通过使用氯膦酸盐脂质体诱导单核巨噬细胞凋亡,可减少VMC及EAM小鼠心脏单核巨噬细胞浸润,减轻心脏炎症和纤维化程度,然而,单核巨噬细胞的凋亡使VMC小鼠急性期心脏病毒滴度增加,死亡率上升[19-20],这表明清除所有单核巨噬细胞在减轻心脏炎症的同时,可能损害了它们的抗病毒能力。因此,研究者在EAM小鼠体内使用靶向CCR2的siRNA,抑制CCR2-CCL2通路介导的炎症单核细胞运输,结果发现,CCR2 siRNA的使用阻止了单核细胞前体从骨髓中的释放以及脾脏单核细胞向心脏中的募集,减少了心脏巨噬细胞的浸润,并减轻了炎症和纤维化程度[21]。同样,研究者通过在VMC小鼠体内使用CSF-1 siRNA使骨髓来源的单核细胞的发育和增殖被抑制,发现心脏单核巨噬细胞的浸润降低,心脏损伤减轻,同时病毒的清除不受影响[22]。而相反,敲除小鼠体内趋化因子受体CX3CR1,可抑制Ly6Clow单核细胞的募集,阻碍CCR2-巨噬细胞的发育和补充,导致CVB3感染的小鼠炎症细胞浸润增多,心肌损伤和纤维化加重[23]。因此,特异性靶向炎性单核细胞向心脏的浸润是减轻心脏炎症的有效手段。
3.3 抗原提呈和适应性免疫
单核巨噬细胞可调控自身反应性T细胞的活化和分化,参与适应性免疫(图1)。自身反应性T细胞的启动是VMC免疫损伤和进展成为DCM的关键原因。研究表明,Ly6Chigh单核细胞可以进入心脏并获得抗原,并随后迁移至脾脏或淋巴结,诱导T细胞的成熟和增殖[24]。同时,在心脏的单核细胞分化为巨噬细胞,吞噬坏死心肌并将其分解为多肽与MHC Ⅱ分子结合,呈递给T细胞,部分高表达CD11c的巨噬细胞被称为“单核来源的树突状细胞”,有更强的处理和呈递抗原的能力,显著诱导T细胞激活[3, 25]。巨噬细胞表面的共刺激分子CD40、CD80和CD86提供第二信号,增强T细胞的活化和增殖[25]。单核巨噬细胞分泌的细胞因子调控T细胞的分化,使其转变为Th1、Th17或Treg表型。单核巨噬细胞是IL-12的主要来源之一,IL-12通过STAT4信号诱导Th1细胞分化,促进EAM的发展[26]。用抗体阻断人类单核细胞上的TLR2或多肽结合表位,可降低其Th17相关细胞因子IL-1β、IL-6、TGF-β、IL-23的分泌,从而抑制体外Th17的活化[12]。氯沙坦作为一种AngⅡ受体拮抗剂,可通过抑制AngⅡ激活的Erk1/2或p38-Stat3通路,降低M1型巨噬细胞和Ly6Chigh细胞的比例,并阻止M1型巨噬细胞诱导的Th17细胞的分化,从而有效改善EAM的进展[17]。而过继转输M2型巨噬细胞至心肌炎小鼠体内,可通过IL-10和TGF-b的分泌提高小鼠脾脏Treg细胞的比例,减轻自身免疫反应[15]。因此,通过改变单核巨噬细胞的表型和功能,可调控T细胞介导的心脏自身免疫反应。
3.4 组织修复和纤维化
单核巨噬细胞调控心肌炎后期的组织重构和纤维化(图1)。在心肌炎的慢性期,心脏浸润的Ly6Chigh单核细胞会转变为Ly6Clow巨噬细胞,同时Ly6Clow单核细胞浸润至心脏并分化为巨噬细胞[27],这些巨噬细胞以M2表型为主,分泌多种细胞因子介导损伤修复:精氨酸酶1、TGF-β和IL-10诱导肌成纤维细胞产生胶原蛋白,血管内皮生长因子诱导血管生成,基质金属蛋白酶和基质金属蛋白酶组织抑制因子调节细胞外基质网络。而M1型巨噬细胞分泌的TNF-α和IL-1会刺激成纤维细胞分泌过量的细胞外基质,导致基质沉积和纤维化不良[28]。淋巴细胞分泌的IL-17诱导成纤维细胞分泌GM-CSF,阻碍了Ly6Clow单核细胞向修复型巨噬细胞的分化和增殖,造成了炎症的持续和纤维化加重[7]。同样,敲除N4a1基因使Ly6Chigh单核细胞转变为Ly6Clow表型受阻,引起心脏愈合不良,纤维化增加和心力衰竭[27]。相反,肌成纤维细胞诱导M1型巨噬细胞的凋亡,并使巨噬细胞向M2型转变,促进炎症的消退[29]。同样,在EAM急性期体内使用脂质体,可促进小鼠Ly6Chigh单核巨噬细胞转化为Ly6Clow表型,减轻疾病后期心脏纤维化,恢复心脏功能[20]。因此,调控单核巨噬细胞的表型可影响心脏的修复和纤维化,改变晚期DCM和心力衰竭的进程。
4 小结与展望
以上研究说明,单核细胞和巨噬细胞在心肌炎最初的病毒清除,到中期的自身免疫和炎症损伤,再到后期的组织修复和纤维化中发挥了多种效应功能。然而,单核巨噬细胞的起源、表型和功能似乎比以往认为的更复杂,还有几个方面有待阐明:心脏浸润的单核巨噬细胞是否包含更加复杂的功能群体?哪种单核巨噬细胞亚群对心肌炎发展最为重要?单核巨噬细胞如何与心脏其他细胞对话?明确以上问题,都将进一步提高我们对心肌炎的了解,帮助我们找到更加有针对性的治疗方法。
利益冲突:无
猜你喜欢
祝您健康(2024年1期)2024-01-11 04:39:32
长春中医药大学学报(2017年1期)2017-04-16 05:56:51
中国继续医学教育(2015年4期)2016-01-07 07:38:10
首都医科大学学报(2015年4期)2015-12-16 13:00:08
现代检验医学杂志(2015年5期)2015-02-06 01:42:11
温州医科大学学报(2014年7期)2014-07-18 02:43:14
无机化学学报(2014年12期)2014-02-28 17:33:53
无机化学学报(2014年7期)2014-02-28 17:32:11
现代检验医学杂志(2014年5期)2014-02-02 02:51:34
食品科学(2013年23期)2013-03-11 18:30:07
