
新能源化工热力学
2021-09-28 03:37练成程锦黄盼陶浩兰杨洁刘洪来
化工进展 2021年9期
练成,程锦,黄盼,陶浩兰,杨洁,刘洪来,3
(1 华东理工大学化学与分子工程学院,上海 200237;2 化学工程联合国家重点实验室,上海 200237;3 华东理工大学化工学院,上海 200237)
能源存储与转换是目前人类能源相关技术中的重要课题,主要涉及电化学过程,因为电是一种品位很高的能量,可以通过其他形式能源与电能的互相转换来高效地利用能量,在一定程度上解决能源问题。电化学是研究两类导体(电子导体,如金属或半导体;离子导体,如电解质溶液)形成的接界面上所发生的带电及电子转移变化的科学,主要涉及能量的储存、释放和转化过程。其高速发展离不开热力学的研究指导。
热力学主要分为两大部分:一个部分为实验方面对物质热力学性质的研究与热力学数据的测定;另一个部分为理论计算方面对物质性质的模拟与预测,从理论的角度阐明物质能量转换和状态变化的规律。两个部分相辅相成,各有所长。实验是一切化学研究的基础,可以得到所需体系准确的热力学参数,而理论研究则在实验的基础上探索规律与原理。实验研究虽然十分重要,但却有多种限制因素,例如人们不可能测定所有体系的所有热力学性质,也难以对微观的体系进行实验研究。而理论研究则可以通过计算快速对所需研究体系的性质进行预测,也可以通过已知的理论工具去探索实验所无法涉及的领域,如纳米尺度的粒子行为、电子结构对体系的影响。因此,本文主要从理论计算方法的角度介绍热力学在电化学能源存储与转换的应用。化工热力学通过对电化学过程、材料、工艺的研究以及计算机技术的辅助,可以快速且定量地解决大量关键问题。除了传统的经典热力学以外,近年来分子与统计热力学也有着蓬勃发展,而今的研究已经更关注非平衡态热力学过程。最后,计算机技术的发展也为热力学研究带来了高通量计算与机器学习的方法。综上所述,热力学研究大幅度提高了电化学领域的发展速度。
本文整理、分析了理论计算方面热力学在电化学能源存储与转换领域的相关重要理论与最新研究成果。对不同热力学理论方法的研究进行分类和介绍,并针对其中分支的经典热力学、统计热力学、非平衡态热力学、机器学习等关键领域进行了详细探讨,在文末分析了今后发展中应重点思考和解决的问题,以期为电化学中热力学的进一步研究与电化学行业的进步提供参考与启发。
1 电化学的经典热力学
1.1 经典热力学概述
热力学在形成初期只是研究热能与机械能间的转换,后来逐渐发展为研究与热现象有关的各种能量转换和状态变化的规律。经典热力学研究内容主要以热力学三个定律为基础,利用热力学数据,研究平衡系统各宏观性质之间的相互关系,揭示变化过程的方向和限度,结论具有普遍性。但是它不涉及粒子的微观性质,不能阐明体系性质的内在原因,不能给出微观性质与宏观性质之间的联系,不能对热力学性质进行直接的计算。要克服这些缺点必须从分子的微观结构和内部运动去认识体系及其变化。
经典热力学对电化学的研究也主要集中在宏观性质方面。随着电化学工业规模的扩大、新过程的开发以及计算机的使用,经典化工热力学的研究和应用也有了较大的发展。
1.2 经典热力学在电化学能源存储与转换领域的应用
电能是一种经济、实用、清洁且容易控制和转换的能源形态,如何将其他类型的能源(风能、潮汐能、太阳能等)转化成电能并存储,这一过程与电化学息息相关。传统电化学主要研究电能和化学能之间的相互转换,如电解池和原电池。
经典热力学主要解决宏观的稳态问题,对于宏观电化学过程与液态电解质溶液的描述已经有了成熟的研究。本节将着重于介绍三个方面的热力学研究:电化学过程、电解质溶液、电解质溶液的过量函数(活度系数)模型。
1.2.1 电化学过程的热力学
从形式上来说,电化学过程与一般化学反应的区别在于:反应在电极的表面上进行,溶液中有离子的迁移,并有电能的输入和输出。从热力学原理分析,最显著的特征是各相电位不同,过程进行时不仅做体积功,还伴随电功。
一般恒温恒压下的化学反应只涉及体积功,dGT,p,dW′=0≤0,其中G 为吉布斯自由能,反应中吉氏函数必定减少。
(1)两种电化学过程 在电化学过程中,系统对外输出电功或环境对系统输入电功,采用ΔGT,p≤W′ele,根据ΔGT,p大于零还是小于零,可分为两类。
(a)ΔGT,p< 0的过程,这类过程可以对外输出电 功, W′ele< 0, W 为 电 功, 最 大 可 输 出W′ele= ΔGT,p。这种电化学过程是化学电源的基本过程。
(b)ΔGT,p> 0的过程,这类过程必须得到电功才能进行,W′ele> 0,最少应得W′ele= ΔGT,p。这种电化学过程是电解和电冶炼工业的基本过程。氯碱、电解铝、电解镁等工业的基本过程都是ΔGT,p> 0的过程。
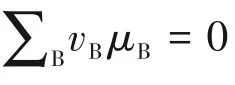
(3)电池的电动势 电池建立后,各相即存在一定的电位,界面两边就有一定的电位差。当没有外接负载,在开路下进行一微元过程时,电动势就是开路下电池中界面电位差的代数和。
不论是化学电源还是电解,当外加电压与电动势绝对值相等,但方向相反时,所进行的电化学过程为可逆过程。

最后,介绍电化学过程的有效能。电功可以任意地转换,是品位最高的能量,因此电功即可定义为有效能。过程热力学分析原理不变[1]。
1.2.2 电解质溶液的热力学性质
电解质溶液的最大特点是溶液中不仅存在分子,如溶剂分子和未离解的电解质分子,还存在由电解质离解产生的带正电荷或负电荷的离子。根据电解质在溶剂中的离解能力,理论上可分为两大类电解质溶液。第一类电解质溶液中的电解质是完全离解的,没有未离解分子或正负离子缔合物,如碱金属、碱土金属、过渡金属的卤化物以及一些过氯酸盐的水溶液。第二类电解质溶液中的电解质则是部分离解的,溶液中有未离解的、原子间按共价键形式结合的分子,如各种强酸和弱酸、弱碱的水溶液;或有正负离子缔合形成的离子对,如二价金属的硫酸盐以及一些强碱的水溶液等。


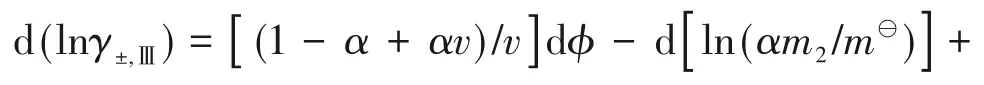

经典热力学方法,还可得到其他过量函数。
1.2.3 电解质溶液的过量函数(活度系数)模型
随着现代统计力学理论在电解质溶液中的应用,电解质溶液理论取得了长足发展,建立了多个在工程计算上有广泛应用的模型[1-6]。

(2)Pitzer 模型 Pitzer 将离子处理为带电硬球,并引入经验的维里展开,以计及非静电相互作用。各种电解质的参数已被列出[2-3]。参数与温度关系[7-8]已被研究,可用于混合电解质溶液[9]。陆小华、王延儒和时钧等[10-12]对Pitzer 参数以及对混合溶剂的推广进行了深入研究。

(4)电解质溶液的积分方程理论和微扰理论 电解质溶液模型的最新发展主要是在统计力学的积分方程理论和微扰理论的基础上发展实用的工程计算模型,多数是在Blum的平均球近似(MSA)结果的基础上研究电解质溶液。
Blum采用MSA简化OZ积分方程中的离子间直接相关函数与离子间位能函数的关系,对具有不同大小和不同电荷的带电硬球混合物(原始模型)获得了解析式。其过量亥氏函数可表达为离子间硬球排斥作用的贡献和离子间静电相互作用的贡献两项之和。硬球排斥作用的贡献采用Mansoori-Carnahan-Starling-Leland[20-21]状态方程计算。离子间静电作用的贡献由Blum[22]的MSA 计算。在Blum的MSA 理论基础上可以进一步考虑离子间的其他相互作用,从而改进模型的计算精度[23-25]。
2 电化学的分子和统计热力学
2.1 分子和统计热力学概述
分子和统计热力学是在经典热力学与统计物理学的基础上发展起来的。在经典热力学领域,人们已经可以解决大部分宏观问题,但是由于经典理论的局限性,无法解决一些微观的、复杂的体系。为了解决这个问题,逐渐发展了分子和统计热力学。分子热力学在分子水平上研究工程或应用中出现的热力学问题,起源于玻尔兹曼等从分子间相互作用力的角度出发对统计热力学的研究,其主要内容是提供表达分子结构的分子参数与热力学性质间的定量关系。分子和统计热力学在经典热力学的基础上,开始关注单个粒子的贡献与分布,在理论上引入了与粒子位置(r)有关的函数,也开始考虑了量子化学中电子的影响。分子和统计热力学在长期发展中,主要诞生了以下强有力的研究工具:基于量子力学的密度泛函理论(QDFT)、经典密度泛函理论(CDFT)、联合密度泛函(Joint DFT)、基于反应力场的分子动力学(ReaxFF MD)和从头算分子动力学(AIMD)。分子和统计热力学将人们探索热力学的领域又一次拓宽了。
2.2 分子和统计热力学在电化学能源存储与转换领域的应用
电化学的发展中诞生了许多新的问题,经典热力学已经无力解决,需要新的方法、新的思路来进行突破。分子和统计热力学的理论在过去几十年里已经突飞猛进,在电化学能源存储与转换领域可以解决大量问题,但以稳态为主,下文主要介绍五个分子和统计热力学理论方法及其所解决的问题:①量子力学的密度泛函理论(QDFT),可以研究某些难以解释的实验现象及其相关机理;②经典密度泛函理论(CDFT),探究粒子在带电表面(孔道)、受限空间中的分布;③联合密度泛函理论(JDFT),自洽高效地解决复杂固液表界面问题;④分子动力学(MD),可以分析机理、筛选材料,还可以获得原子的动力学轨迹来研究扩散机制等。
2.2.1 量子力学的密度泛函理论(QDFT)
随着科技的发展,大量的新型材料应需而生,而在应用过程中某些实验现象难以解释及其相关机理尚不明确[26]。例如,新材料为何能提高电池性能;电池中锂枝晶形成,电解质分子分解和SEI膜形成的机理;催化过程中催化反应机理等。针对这些问题,需要从材料电子结构的角度出发去寻求突破,而传统的经典热力学无法处理这样复杂的问题。因此,量子力学的密度泛函理论(QDFT)作为一种重要的方法,可用来描述并定量计算该热力学问题。
QDFT 求解体系的电子结构和能量信息最初的思想是通过Schrödinger方程求解体系的波函数来获得[27-28],如式(1)。

但是实际体系非常复杂,要严格求出Schrödinger方程是不可能的,所以必须在物理模型上作一系列的近似和假定才能求得多电子体系中的电子结构。因此,Born 和Oppenheimer 等[29-30]提出原子核质量比电子的质量大得多,电子运动的速度远远大于原子核运动的速度,因此可以把原子核认为是近似不动的点电荷,可以根据Born-Oppenheimer 近似把体系的波函数分离成电子波函数和核波函数的乘积。随后,经过Hartree、Fock和Slater[31]对方程进行改进提出了Hartree-Fock 方程,如式(2)。




其中

在求解Kohn-Sham 方程时候必须知道泛函和交换相关项即第三项的关系,交换相关项分为交换能和相关能。目前来说,没有得到交换相关项的准确函数表达形式,但是得到了非常实用的近似泛函形式,其中主要有局域密度近似(LDA)泛函[34]、广义梯度近似(GGA)泛函[35-37]。
QDFT 以Hohenberg-Kohn 定 理 以 及Kohn 和Sham 所提出的方程为基础,经过几十年的发展,在物理、化学和材料等领域被广泛的应用。通过QDFT 方法,电化学中吸附能和扩散活化能的计算、锂枝晶形成的机理等问题都可以迎刃而解。
在储能材料领域,主要问题体现在对吸附能和扩散活化能的计算上。众所周知,作为一个良好的储能材料需要对带电离子(锂离子、钠离子等)具有良好的吸附性能,并且也要有利于这些带电离子在材料上的扩散。良好的吸附和扩散性能体现在能量上就是需要大的吸附能和低的扩散活化能。Tang等[38]利用QDFT 计算了锂离子在Ti3C2以及Ti3C2X2(X=F、OH)上的吸附能和扩散活化能,如图1 所示。结果表明,在Ti3C2上有官能团修饰时,是不利于离子的扩散的。Tang 等通过计算认为,Ti3C2是锂离子电池中良好的储能材料,但是当表面有官能团(—F 或者—OH)修饰时,电池的储锂性能将会下降。因此,实验中应该避免Ti3C2表面—F、—OH 等官能团的修饰。Zhou 等[39]通过QDFT 计算了Li2S6在不同金属硫化物材料(Ni3S2、SnS2、FeS、CoS2、VS2、TiS2)上的吸附能以及锂离子在这些材料上的扩散活化能,结果如图2所示。他们的结果显示Li2S6在VS2、TiS2和CoS2上的吸附能较大,并且锂离子在这些材料上的扩散活化能较小。因此,他们预测VS2、TiS2和CoS2作为电极材料可以提高锂硫电池的性能,并且通过实验证实了他们的观点。

图1 锂离子在Ti3C2、Ti3C2F2和Ti3C2OH2上扩散的三条路径和对应的扩散活化能[38]

图2 Li2S6在多种材料上的吸附能与锂离子在多种材料上的扩散活化能[39]
QDFT 还可以研究锂枝晶形成的机理。本文作者课题组先前的研究[40]通过QDFT 证明成核是锂晶体在理想界面上生长的关键。位错缺陷降低了第一个锂原子的吸附能,并对锂晶体的生长产生了长期的影响。对于杂原子缺陷,如图3所示,Cl原子漂浮在锂晶体上,可以抑制锂枝晶的生长。

图3 使用QDFT进行几何优化前后Cl原子的位置发生变化[40]
QDFT 除了得到能量方面的信息外,还可以得到例如态密度(DOS)、电荷密度、差分电荷密度和轨道等电子结构方面的信息。这些信息对储能材料的筛选也有很大的帮助[38,41-43]。
2.2.2 经典密度泛函理论(CDFT)
电化学相关材料表界面平衡及非平衡态性质是一个重要的热力学课题,探究粒子在带电表面(孔道)、受限空间中的分布对于电化学系统的设计有重要意义。为了精确描述并定量计算,在传统半经验分子热力学统计方法的基础上,发展出了经典密度泛函理论(CDFT),其可保留宏观系统的微观体系;而相比分子动力学模拟,具有更高的计算效率。
经典密度泛函理论是基于Hohenberg-Kohn 定理[32]建立的,定理指出,在体积、温度和流体化学势(μ)一定的开放体系,即巨正则系综中,每种分子的潜在化学外势ψi(r),只由体系平衡时的密度分布ρi(r)所决定。对于单组分体系,内在Helmholtz自由能可表示为式(5)。

其中,A为Helmholtz 自由能。内在Helmholtz自由能与巨势能的关系可表示为式(6)。

内在Helmholtz 自由能以及巨势能是粒子/分子的单体密度分布的函数,当巨势能取极小值时,可获得系统平衡态的微观结构和宏观热力学性质,如式(7)。

拓展到多组分流体,巨势能和流体的内在Helmholtz自由能可通过勒让德(Legendre)变换互相关联,如式(8)。
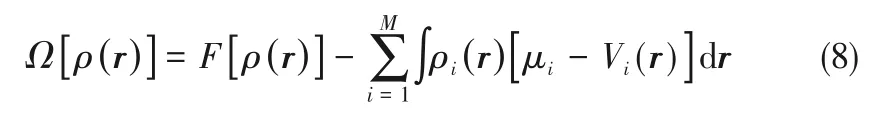
ρ(r)=[ρ1(r),ρ2(r),⋅⋅⋅,ρM(r)],其中ρi(r)、μi和Vi(r)分别为第i种粒子的密度分布、化学势和外势的作用能。
系统的内在Helmholtz 自由能可分为两部分:理想气体Helmholtz 自由能Fid以及来自分子间相互作用力的过剩项Fex,如式(9)。

对于多组分流体,理想气体Helmholtz 自由能通过统计力学推导,可表示为式(10)。

进一步可得到非均匀流体理想气体的内在Helmholtz自由能,如式(11)。

精确表示过剩Helmholtz自由能对于CDFT计算至关重要,然而确切公式通常是未知的。因此只能采用各种近似,以描述流体分子间复杂的相互作用。
若能得到剩余自由能对密度分布的泛函表达式,将式(8)~式(11)代入式(7)可得描述流体密度分布的Euler-Lagrange方程。
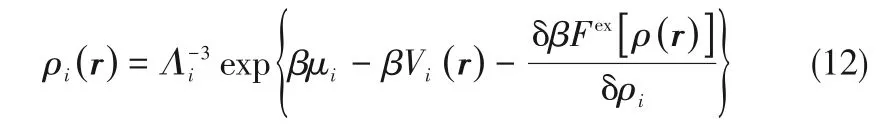
目前主要有两种方法用于计算体系的过剩Helmholtz 自由能:一是泛函密度展开;二是将过剩Helmholtz自由能分解[44-46]。
得益于CDFT的准确性和高效率,它已成为研究超级电容器电极中双电层结构和电化学性能的理想工具。Jiang 和Wu 等[47-48]利用CDFT 预测了带电平板表面的离子液体分布情况。如图4所示,带电平板表面吸附反离子(与平板带电性相反),形成带电量相反的紧密层,进而形成共电极材料离子层(与平板带电性相同)。同理,依次形成阴阳离子层交错的结构,这也与实验及其他模拟方法如MD[49-50]、MFT[51]、MC[52-53]的预测结果保持一致。

图4 离子液体电解液在带电平板表面的分布(ρion/ρbulk)随位置(z)的变化
求解电极材料表面附近双电层的结构后,根据下列公式可计算不同外电势下电极材料中储存的电荷总量Q、微分电容CD和积分电容C,如式(13)。

在此基础上,通过研究表界面性质,还可以探究电极材料[54-58]与电解液[59-61]对于超级电容器性能的影响。
2.2.3 联合密度泛函理论(JDFT)
为了进一步提高电容器等储能设备的电化学性能,多种新型电极及电解质材料已被广泛应用。此时,固液界面环境十分复杂:当电极界面不能假设为理想的金属或硬壁边界时,电极化学将起到十分重要的作用。直接利用量子密度泛函理论处理整个体系,计算成本高,现阶段难以实现。因此,Arias等[62]提出联合密度泛函理论(JDFT),以自洽高效地解决固液表界面问题。
JDFT 利用QDFT 处理溶质,同时利用CDFT 处理溶剂。通过最小化总的自由能,可得到平衡状态下的固液界面性质。JDFT 中自由能可表达为式(14)。

式中,AHK是Hohenberg-Kohn Theorems 提出的电子能;Alq是经典密度泛函表示的液体自由能[63];ΔA是源于溶质与溶剂相互作用的耦合能量。
对于双电极系统,工作电极的电势μ定义为单个电子从工作电极(电化学势μW)迁移到参比电极(电化学势μR)的能量,即ε = μR- μW。如果选定参比电极的电势为0,即ε =-μ。当电极在模拟中为电中性时,μ 和零电荷电势(PZC)有关。通过对比理论预测和实验标准的PZC,可得到标准氢电极的电势。随后即可作出不同表面电荷下电极电势曲线,通过微分计算可得到电极电容。
以超级电容器中广泛应用的石墨烯材料为例,与传统碳电极不同,电容来自于双电层电容以及量子电容两部分的贡献。前者来自带电界面的电子-溶剂排斥作用,将影响双电层的结构和屏蔽电位。后者来自额外的电极电势降,当电极带电时,电子开始占据较高能态,因此降低了整体电容。通过将经典模拟中的双电层电容与电子结构中的法拉第电容相结合,可以计算整体电容,如式(15)。

对于2D 材料,量子电容和双电层电容相当,量子电容对总电容的影响不可忽略。Zhan等[64]利用JDFT 研究了石墨烯层数对量子电容及总电容的影响。JDFT 还可用来考察石墨烯超级电容器的介电屏蔽[65]、边缘效应[66]甚至是赝电容器的充放电机制(如RuO2[67]、MXene[68]),由此获得更加真实的电极材料性质及其与电化学性能间的构效关系。
JDFT 也被证明是金属电池材料表界面性质研究的良好工具,在原子层面上理解电极电解质界面的物理过程对于设计改性电池十分重要。金属材料如锂、钠具有高能量密度的特点,已被广泛应用为电池阳极材料。然而它们在充电时会局部成核,并形成针状结构,阻碍了其商业化发展与应用。Gunceler等[69]利用JDFT预测电解质中卤化物阴离子(特别是氟离子)的存在会使得Li+在锂电极电解质界面扩散的活化能垒降低0.13eV。这可将表面扩散率增加两个数量级以上,大幅改善锂电沉积的稳定性并抑制液体电解质中枝晶的生长,随后的实验也验证了这一猜想[70-71]。Ozhabes等[72]更深入地在原子水平上探索了卤化物阴离子抑制枝晶的机制。
由于JDFT 准确高效地解决了固液界面问题,已在储能设备、燃料电池领域的机制探究上取得重大突破。随着相关理论基础的进一步完善,JDFT有希望指导更多电化学相关材料的设计优化。
2.2.4 分子动力学(MD)
能量的储存、释放和转化往往伴随着微观行为(如扩散、吸附、化学反应等)的发生,想要更深入地认识这些过程,就需要使用分子模拟从原子尺度上进行更加细致的研究。利用QDFT等量子化学计算手段,可以在原子、电子层面研究体系中的各种微观性质;利用CDFT、DDFT等半经验的统计热力学方法,可以将微观性质与介观、宏观的现象进行结合。前者具有很高的可靠性和精确性,但无法直接对动态过程中的现象进行描述;后者虽然能够预测介观、宏观上的现象,但无法表现粒子的具体行为。
为解决上述问题,分子动力学(MD)应运而生。发展最成熟、应用最广泛的分子动力学方法是力场分子动力学(FFMD),但其无法描述化学反应,对未知或复杂的体系也难以作出可靠的模拟,因此发展出了两种更为先进的分子动力学方法:基于反应力场的分子动力学(ReaxFF MD)以及从头算分子动力学(AIMD)。
(1)力场分子动力学(FFMD) 分子动力学方法是Alder 和Wainwright[73]在1957 年开创的,他们提出了基于刚球势的分子动力学方法。Rahman[74]于1964 年利用Lennard-Jones 势函数对液态氩的性质进行了模拟,Rahman 和Stillinger 在1971 年模拟了具有分子团簇行为的水的性质,1977 年Rychaert 和Ciccotti 等提出了约束动力学方法。此后,人们对这种方法作了很多方面的改进,尤其是1980 年后,随着计算机技术的快速发展以及多体势函数的提出和发展,更加加速了分子动力学模拟技术的发展运用。
具体来说,分子动力学模拟是以分子(原子或其他离子,为方便描述故统称为分子)作为基本的研究对象,把待模拟系统看作大量分子的集合,运用经典力学或者量子力学的方法来研究这些分子的运动规律,从而最终得到体系的宏观性质和基本规律。分子动力学首先要选取合适的力场。力场是势能函数的经验表达式。分子的势能是各类型势能的总和,包括非键结势能、键伸缩势能、键角弯曲势能、二面角扭曲势能、离平面振动势能和库仑静电势能。自分子动力学模拟方法开创以来,计算所使用的力场也一直在发展,研究者针对不同应用目的发展出各种不同的力场,所以不同的力场各有其优点和适用范围,这也就决定了在应用分子动力学模拟时要根据模拟的系统选择最合适的力场。
在电解过程中,由于电极表面吸附的水的参与,广泛用作电解质的湿润疏水性离子液体的电化学窗口减小了。Chen等[75]使用FFMD探讨在湿润离子液体中添加锂盐对电极表面吸附水的影响,结果表明,通过加盐,大部分水分子被从阴极和阳极排斥开。该工作提供了潜在的机制和一个简单但实用的方法来保护潮湿的离子液体免受电化学性能退化。
有机溶剂/水混合电解质是一种很有发展前途的新一代储能装置,是工业电气化和脱碳的关键。然而,人们对纳米多孔超级电容器电极中电解质离子的扩散行为知之甚少。Li等[76]通过实验和数值模拟对超级电容器的电阻和离子动力学进行了系统的研究。在纳米多孔电极上的电化学结果显示,超级电容器电阻随溶剂迁移率的增加呈非单调(先减小,后增大,再减小)的趋势,这对长期以来认为超级电容器电阻随有机溶剂迁移率的增加而减小的观点提出了挑战。通过对0.95nm纳米通道内电解质离子扩散的分子动力学数值模拟,验证了这种反常趋势。以上进一步证实了溶剂分子的范德华尺寸和孔道宽度在确定纳米多孔电极电解质电导率中的关键相互作用。
Ma等[77]应用FFMD来描述具有多孔碳电极的咪唑离子液体,建立了包括狭缝和圆柱孔在内的基本几何模型,以确定离子在充放电过程中的行为。通过研究局部电荷的变化,表明电极的电荷或极化与离子动力学或弛豫有关,特别是在电极附近。探索了实现循环伏安法和电化学阻抗谱技术的途径。
氧化石墨烯(GO)纳米通道中的离子液体(IL)流动对基于IL 和GO 的流体装置和其他化学分离技术的性能起着关键作用。Wang 等[78]通过分子动力学模拟研究了离子液体在GO纳米通道中的流动行为,确定了滑移速度与剪切应力之间的定量关系,证实了GO附近IL层结构的变化是界面性质发生巨大变化的原因。此外,由于受限离子液体库仑有序的部分破坏,黏度会随着羟基化程度的增加而增加。同时,由于IL/GO界面具有较强的相互作用网络,羟基对IL流动的影响比水在GO纳米通道中的影响更为显著。发现羟基化是一种令人信服的方法来调节IL在纳米通道中的流动。
(2)基于反应力场的分子动力学(ReaxFF MD) 大规模ReaxFF-MD 模拟得到的原子轨迹和键级信息中蕴含着复杂的化学反应信息,便于进行人工分析机理。基于反应力场的分子动力学模拟(ReaxFF MD)在化学物理学、材料科学以及生物物理学发挥着举足轻重的作用。ReaxFF 反应力场的方法已经成功地应用于一些反应动力学模拟的研究中,应用体系包括碳氢有机小分子体系、高分子体系、高能材料体系、金属氧化物体系以及过渡金属催化剂体系[79-80]。
ReaxFF 反应力场是van Duin 及其同事开发的[81]。反应分子动力学(ReaxFF MD)是一种极具潜力可应用于模拟较大规模分子体系(>10000个原子)的化学反应的新方法。
ReaxFF系统能量描述如式(16)。

式中,各项分别为孤对电子能量修正项Elp;过配位能量校正项Eover、Eunder;价角能量项Eval;键角能量惩罚项Epen;四体共轭项Econj、Etors;三体共轭项Ecoa;C2修正项EC2;范德华作用项EvdWaals;库仑作用项ECoulomb;氢键作用项EH-bond。
研究者利用ReaxFF MD 方法可以探究锂离子在电解液中的存在形态与扩散情况、SEI膜的形成机理与结构、锂离子在电极之间的传输方式等。Hossain 等[82]将ReaxFF 反应力场参数集扩展到有机电解质种类,例如碳酸亚乙烯酯、碳酸亚乙酯、LiPF6盐以及碳酸乙基甲基酯;并利用该力场研究了锂离子和锂原子分别对于锂溶剂化的反应机制。该工作对初始充、放电循环期间电解质分解的机理进行了阐释。
对于锂离子电池阳极,电极材料的高锂容量,稳定的循环性能一直是人们研究的热点。
从图5(a)中可以看出,硅的理论容量高达4200mAh/g。因此,硅-氧-碳杂化纳米结构作为锂离子电池的有希望的负极材料受到了广泛关注。

图5 基于反应力场的分子动力学示意图[83-85]
Han 等[83-84]在Si/C/O/Li 反应力场开发方面做出许多重要工作。报告了不同种类的硅阳极(原始硅和SiOx),各种碳酸盐电解质的不同类型和组成以及添加剂对SEI形成的影响。随后,该团队利用该力场研究了sp2 碳包覆的Si 和SiOx纳米结构(例如纳米线(NWs)和纳米颗粒(NPs))的原子锂化行为。该工作为Si-O-C 混合阳极的设计提供了理论指导意义。具体工作如图5(b)所示。
MXenes 最近发现的二维(2D)材料,由于其出色的电导率,可作为超级电容器电极材料,这引起了研究者极大的兴趣[85]。通过ReaxFF-MD 分析了位于9个不同温度(200K、250K、260K、270K、280K、290K、300K、350K、400K) 下 的 双 层MXene层之间的单层水中的质子迁移,对三个独立创建的MXene-水模型的仿真中,发现质子扩散常数随温度升高而变化的异常趋势。如图5(c)所示。
(3)从头算分子动力学(AIMD) 经过数十年的发展,人们已经针对多种原子、分子(或基团)、粗粒化粒子和不同体系建立了分子力场,但远未能完全满足分子动力学研究的需要,对于复杂、未知的体系往往难以获得可靠的结果。此外,力场分子动力学的模拟结果完全取决于力场参数,而这些参数来自于实验测定和量化计算的结果,只能体现原子、分子层面的特征信息,如果行为受电子层面的性质变化影响很大,力场分子动力学将难以描述。
从头算分子动力学(AIMD)是一种使用量子化学方法计算原子的受力情况,可以从电子层面出发描述粒子的动态行为,其计算基本流程如图6所示。因其基于量化计算,具有精度高、可靠性好、普适性强的优点,并且可以对化学反应的本征过程进行表现。但是,AIMD 的计算成本更加昂贵,在相同体系下的时间成本相对于FFMD 高出几个数量级[86]。

图6 AIMD模拟的基本流程
在纳米尺度的约束下,流体中的粒子表现出与在体积(Bulk)区完全不同的行为。利用AIMD,能够以高的准确性和可靠性对粒子的反常扩散和传输进行动态研究[87-91]。
Mo 等[89]率先在锂离子导体材料的离子扩散研究中应用AIMD 模拟,自此以后AIMD 作为一种研究快速离子导体的标准技术被用于Na+、Mg2+、O2-等很多种离子的研究中[92-94]。AIMD(以及经典MD)的优势在于,材料中的扩散行为直接从原子的动力学轨迹中观测,而不是人为指定了扩散机制。对于复杂材料(如超离子导体)而言,其扩散机制的猜测十分困难,且无法保证猜测的可靠性,AIMD 模拟在发现和识别不同体系的迁移机制方面发挥了独特作用,如最近对超离子导体的研究发现,锂石榴石[91,95]等中的迁移行为是多离子协同的结果,而非孤立离子的跳跃。此外,AIMD 模拟可以对扩散行为影响因素的贡献进行采样和统计,这对建立该体系中扩散机制的数学模型有极大的帮助。
目前,AIMD 模拟已经成为研究扩散机制和特定材料的扩散特性时广泛使用的手段。使用AIMD模拟来提取扩散特性需要解决三个问题:①以最小误差从模拟结果中提取扩散特性;②将提取出的扩散特性的误差进行量化;③可靠的物理范围。为此,He等[96]建立了分析程序,从AIMD模拟结果中提取扩散特性并量化其统计误差,并估计了AIMD模拟可用的温度范围。
3 电化学的非平衡态热力学
3.1 非平衡态热力学概述
目前,以平衡态和可逆过程为基础的平衡态热力学已经形成了完善的理论结构并广泛的用于各种物理、化学过程的研究。然而,平衡态热力学仅研究平衡态及其过程的特殊情况,有很大的局限性[97-99]。热力学在非平衡态过程中的应用有多种形式,最简单的是基于局部平衡假设的线性非平衡态热力学。假定非平衡态过程发生的系统可以被分成一系列小区域,每一个小区域的热力学性质都和平衡态一致。局部平衡假设是否使用于某一系统取决于是否存在两个大小不同的弛豫时间,即是否存在远小于整个体系达到统计平衡的弛豫时间τ的时间τr,使在时间间隔τr内,这些小区域可以建立局部平衡。局部平衡概念适用于各种常见的气体和液体的宏观系统,以及大多数反应过程的碰撞速率小于总碰撞速率的运输过程和化学反应,而不适用于在碰撞非常稀少的高度稀有气体。另一种众所周知的应用是第二定律分析法,这一分析方法基于Gouy-Sodola定理,说明由于过程的不可逆性,损失的可用能与熵增成正比。熵增的程度被用作与判断过程最优性的标准,主要通过减少过程的不可逆性去减少有用功的耗散和自然资源的枯竭。
3.2 非平衡态热力学在电化学能源存储与转换领域的应用
在电化学相关材料的制备和应用领域,其包含的过程更复杂,如扩散、迁移、传热、流体流动、化学反应,甚至耗散结构主导的过程[100]。这些不可逆过程在储能和转换系统中是高度耦合的,并且随着时间的推移,从非平衡态到平衡态发展。根据线性非平衡热力学,可以用响应矩阵的非对角项来描述不同热力学力对电化学系统中离子输运的耦合效应。解析法侧重于通过数学推导求解唯象系数矩阵,矩阵的非对角项描述不同热力学力的相互作用。分子建模已被广泛应用于研究一些特殊的输运现象,如充电动力学、电动现象和热电效应,这些现象可以提供关于输运过程的详细信息,但需要大量的计算工作。连续性模拟是快速探索电化学系统动态特性的一种有效方法,但它受到稀释溶液理论的限制,使该理论不适用于分子尺度或高外加电位下。而动态密度泛函理论(DDFT)可以在考虑空间效应和其他相互作用的情况下,快速和准确地研究这些运输过程。
如图7 所示,DDFT 是一种综合方法,忽略过量化学势就得到Nernst-Planck (NP)方程。将DDFT与包括泊松方程、Navier-Stokes(NS)方程、热方程和Butler-Volmer(BV)方程在内的其他几个偏微分方程耦合起来可用于描述电化学相关材料制备及应用过程中的多种非平衡态过程。

图7 电解质非平衡热力学的动态DFT理论
Poisson-Nernst-Planck (PNP) 方 程[101-102]既 描述了溶液中物质的质量传递,又描述了电解质中的电势分布,可用于研究与其他过程方程耦合的非平衡过程,如NS方程、热方程和BV方程。
本节基于非平衡态热力学,从传质、反应和传递耦合、热效应三个角度来理解电化学能量储存与转换过程。
3.2.1 非平衡态传质
实际化工过程大多为非平衡态过程,而其中最重要的粒子行为就是传质。不受外力作用时,电解质中的离子会永不停息地做随机运动,而在外电场的作用下,沿着带电表面流动的电解质不仅可以由压力梯度驱动,也可以由电压差产生的电场驱动,形成传质现象,也叫离子迁移。非平衡态热力学在经典热力学、分子和统计热力学的基础上,考虑了时间t的影响,也就是粒子随时间的变化过程。
在某一系统中,假设含有i种与电极接触的离子,zi为对应的化合价,ε为介电常数,n为黏度,T为温度。连续性方程如式(17)。

对于通量,这里考虑扩散和迁移的贡献,首先考虑扩散的贡献,由通用的菲克定律给出,如式(18)。

式中,J是“流”;化学势μ的梯度是热力学“力”,是粒子密度分布,而Γ=D/kBT是迁移率。化学势μ可以分为本征项μint和外场项μext,其中本征项μint的推导与CDFT 类似,取决于系统的Helmholtz 自由能,如式(19)。

其中,F[ρ]= Fid[ρ(r)]+ Fexc[ρ(r)],包括理想自由能Fid和剩余自由能Fexc,理想自由能可以表示为式(20)。


在稳态假设下,通量与从高浓度区域到低浓度区域的梯度成正比,如式(21)。

其中,Di是扩散系数或扩散系数。其次,考虑迁移的贡献,利用爱因斯坦关系,可以用波动耗散引起的扩散系数来表示,如式(22)。

其中,vi指通量速度;E 指系统内的电场。速度是电力(ZieE)与Stokesian 摩擦项(6πηαi,αi是离子半径)之间的比值。
综上,可得到通量的表现形式如式(23)。

这一关系常用于电化学与电化学分析领域。
在电化学能源存储领域,超级电容器可以提供比电池更高的输出功率,并且可以提供比介电电容器更多的能量[104]。由于碳电极包含大量的纳米孔并可被流动性离子液体填充,许多类型的碳基材料已用于电容器电极的制造中。然而,实验测量的超级电容器充电和放电时间尺度与现有的理论预测之间存在很大的差距,并且多孔结构与宏观超级电容器的充电点和动力学之间的关系仍然不清楚。Lian等[103]提出了一个用于解释超级电容器充电动力学的新模型,如图8(a)所示,将电极结构近似为一系列无限薄的平行板堆叠而成,平行板的间隙即典型孔的宽度。模型成功减少了理论预测与实验测量结果之间的时间尺度差距,并可作为扩展突破平面对称性的基础。
锂离子电池作为一种传统的二次电池,依靠锂离子在正负极之间的移动来转换能量,为实现更高的安全性与能量密度,全固态锂离子电池有望成为新一代锂离子电池。全固态锂离子电池的核心组成部分就是固体无极快离子导体。He 等[91]基于密度泛函理论的分子动力学模拟了如图8(b)所示的扩散模型。
在图中所示的扩散模型中,四个流动离子排列在一个由两个单元组成的一维晶格中。整个移动晶格的总能量E 由晶体骨架(即能量景观)的势能φ 和移动离子之间的库仑相互作用给出,如式(24)。

其中,xi是离子i的位置;K是两个流动离子之间的库仑相互作用强度。
该模型阐明了协同离子扩散的低能势垒是超离子导体中独特的移动离子构型和强的移动离子相互作用的结果,为设计具有快速离子扩散的固体材料提供了一般的框架和通用的策略。
半导体光催化技术[图8(c)]能够把丰富的太阳能转化为化学能,是维持地球的未来发展的潜在技术。在半导体光催化过程中,半导体体相内电子和空穴的复合过程比电荷传输和电荷表面催化过程快,这导致半导体光催化剂的体相电荷分离效率降低,严重阻碍了半导体光催化技术的发展与实际应用。Li等[105]发展了一种通过内电场调控来提高半导体光催化剂体相电荷分离效率的全新策略,获得了较高的体相电荷分离效率。

图8 超级电容器及其电极堆叠模型、锂离子电池扩散模型、半导体光催化剂示意图[91,103]
3.2.2 反应和传递的耦合
在非平衡态热力学解决了传质问题后,反应和传递耦合问题也普遍体现在电化学的储存和转化过程中,以备受关注的电催化反应为例,其可持续发展的模式是以太阳能、风能和水力能等可再生的电化学作为动力,通过电化学过程将水、二氧化碳和氮气等小分子转化成高附加值的产品,如氢气、烃类、氧化物和胺,其中涉及析氢反应(HER)、氧还原反应(ORR)、二氧化碳还原反应(CO2RR)以及氮还原反应(NRR)。对于电催化的发展,为了指导高性能电催化剂材料的开发和制备,一个很重要的前沿是利用实验和计算方法快速分析和解释在大量不同类别的催化剂上以及在不同的操作条件(如pH、溶剂和电解质等)下的反应过程和机制[106]。为了研究在催化剂和电解液界面发生的各种传递和反应耦合过程,动态密度泛函理论(DDFT)是一个有效的手段,它是CDFT 在动力学上的延伸,以化学势梯度驱动密度分布[100]。通过很多不同的推导过程都可以得到标准的DDFT 形式,基于唯象理论和线性非平衡态热力学理论的推导方法是其中最快的,它的出发点是通用菲克定律,见式(18)。
由式(17)~式(20)可以获得DDFT 方程的标准形式,如式(25)。

对于化学势μ(r,t),如果只考虑本征项中的理想自由能以及外场项中的外加平均电势,进一步耦合Poisson 方程,可以得到上述的PNP 方程,而表面电极反应可以用BV方程表示,如式(26)。

式中,i0是交换电流密度;αa和αc是量纲为1的阳极和阴极电荷转移系数;n是所涉及的电子数;F是法拉第常数;R是气体常数。这样,可以利用得到的PNP-BV方程去解决电催化体系中的传递和反应的耦合问题,比如通过计算电极尖端的电场、离子分布和电流密度来描述电极尖端曲率对电催化二氧化碳还原的影响[图9(a)][107]。对于电化学系统,理解离子在微结构电极中的传输过程对多孔电极的设计十分重要,Suter 和Haussener[108]利用实验上的电极几何结构构建了一个三维的传质模型[图9(b)],并考虑电解质溶液中的KCO3缓冲反应,以及通过上述的BV 方程表示电极表面的催化反应,来计算CO2还原成CO的过程中局部的浓度分布,进而从优化电极几何和传质的角度提升CO的选择性。
在上述PNP 方程推导的基础上,如果再考虑本征项μint中剩余自由能Fexc的体积排斥项Fhs[ρ],就可以得到修正的通量表达式,如式(27)。

式中,ϕ是电势;a是分子或离子的尺寸。再进一步结合式(17)和Poisson 方程,就得到了修正的PNP 方程。Bohra 等[109]利用通用修正的PNP 方程(GMPNP)描述了电催化CO2还原体系中电极附近的扩散、迁移和反应耦合的现象[图9(c)],其中考虑了因为溶质分子或离子的溶剂化尺寸所引起的体积排阻效应,比较准确地计算了双电层区域的电势和浓度分布,以及双电层结构对CO2扩散的影响。研究传递和反应耦合现象时,外场除了电势外,流体流动的影响也十分重要,Lis 等[110]首次从实验上报道了流体流动对界面反应与EDL 结构的影响,将微流体与表面特性SFG光谱学相结合证明流体流动导致界面水分子的可逆重新排列和浸没表面的表面电荷变化。同样,动态密度泛函理论也可以用于研究流体流动对界面反应的影响,Lian等[111]将经典密度泛函理论与NS 方程以及反应动力学结合来研究溶剂流动与表面化学反应的关系[图9(d)],发现固体表面的溶剂流动对表面反应有显著的影响,进而影响表面电荷密度,因此在高的驱动压力下,流动电流随压力呈现出非线性的关系。

图9 反应和传递的耦合示意图[107-109,111]
3.2.3 热效应
在非平衡态热力学关注的领域,除了主要的传质与反应问题外,热效应也是与几乎所有电化学过程息息相关的,且这个过程是非平衡的,因此前文所述经典化工热力学、分子和统计热力学都无能为力,而非平衡态热力学则可以在一定程度上解决这个问题。热效应主要分为两个方面,即电生热、热生电。
(1)电生热 高温会导致材料老化、内阻增大、电容降低、发生电化学副反应和自放电等问题。而低温会使化学反应速度变慢,电解液阻值变大,锂离子穿越性能变差,活性降低,放电的电流变小,电池的可用容量降低。通过建模可以更好地理解能量转换装置的热行为。
PNP方程假设物理系统是等温的,并使用恒定的温度来描述,这可能适用于那些温度变化很小的系统。然而,电流通常伴随着焦耳热效应,导致相当大的温度变化。d'Entremont 等[112]提出了一种新的双电层电容恒流充放电过程中离子耦合传热的物理模型[图10(a)]。该模型利用修正的PNP模型解释了Stern 层的存在和离子的有限大小。模拟结果定性地再现了文献报道的各种充放电条件下的实验数据[图10(b)]。Hsieh 等[113]利用能量变分方法导出了一个非等温电动力学模型。用变温度下的PNP 方程描述电荷输运,热流满足傅里叶定律。这种Poisson-Nernst-Planck-Fourier模型既满足热力学第一定律和第二定律,又满足Onsager 的倒数关系,因此它是热力学一致的。最后,利用能量法证明了该模型在初始数据很小的假设下的全局适定性。
目前对于双电层形成过程中发生的可逆热效应仍然缺乏统一的认识,充电的热响应也没有得到充分的研究。从热力学的观点来看,在充电过程中,超级电容器的电极熵的表面电荷被反离子电荷屏蔽,因此离子构型熵降低。对于一个准静态带电的绝热超级电容器,热力学要求其满足等熵的条件。离子减少的构型熵被增加的电解质熵(升温)所抵消[图10(c)]。从动力学观点来看,在放电过程中,反离子必须越来越多地扩散回溶液中。这种扩散是在对抗电力时发生的。提高这些离子的电能所需的能量必须从周围的离子和分子中吸收,并从后者的热运动中以动能的形式传递。因此,绝热的超级电容器在放电时降温。Jamssen 等[114]对超级电容器可逆加热的不同观点进行了详细的比较,在定量上调和了可逆加热的两种观点,讨论了热力学观点中的等熵过程的理想气体等熵压缩模型(含亥姆霍兹和古伊-查普曼吸附)和应用麦克斯韦关系式和巨势函数的模型,其中只有后者的结果与动力学的观点中的PNP方程耦合热方程相一致[图10(d)]。

图10 热效应电生热示意图[112,114]
(2)热生电 低品质的热(低于373K)普遍存在于实际生产中,包括来自工业废热流、太阳能加热、地热水库、海洋热梯度、生物质发酵甚至数据中心的废热,是主要的可持续能源。但由于目前的回收技术不具成本效益,因此大部分都被浪费了。热电效应表示温度梯度和电压梯度之间存在直接转换。通过热电效应,可以直接将低品质的热能转换为电能。
温度演化与电流的耦合对理解电解液系统的动力学特性具有重要意义,近年来引起了广泛的关注。Janssen等[115]利用PNP方程耦合热方程研究了电解槽在其两个边界电极的其中一个表面突然施加小的温度升高时的瞬态响应(图11)。通过PNP 模型耦合热方程推导了盐浓度、电荷量和早期(远短于热弛豫时间)以及后期(远长于热弛豫时间)热电压的解析逼近解。对于阴阳离子扩散系数不相等的电解质,热电压表现出两步弛豫过程,与实验数据一致[116]。

图11 热效应热生电示意图[115]
虽然已经成功揭示了温度梯度对离子输运的影响,但目前还没有详细的研究探索纳米通道中热驱动离子输运的调节方法,特别是通过温度差和纳米通道长度来调节的方法。Zhong 等[117]建立了基于PNP方程的计算模型来描述热驱动离子在纳米通道中的输运。研究了不同影响因素对离子输运的影响,包括纳米通道的温差、离子浓度、纳米通道的半宽和纳米通道的长度。结果表明,离子浓度为1mol/m3、纳米通道半宽度为1nm 是理想的用于热电应用的纳米通道体系。
与在体相中对应的热电动势相比,电解质在受限空间里的热电动势有很大的不同。电解质在体相中的热电动势与不同离子的热泳迁移率有关,而即使每种离子的迁移热相同或非常小,电解质在受限空间里的热电动势也可能存在。Dietzel等[118]发现即使在最简单的电解质中,如果被限制在带电的壁之间,也不需要这种Soret 型离子热扩散来诱导热电效应。在约束下的热电动势与通道ζ电位或表面电荷密度成正比。双电层的空间电荷导致了由温度依赖性的电泳离子迁移率驱动的选择性离子扩散,这对于狭窄的通道来说,可能会导致比经典Soret 平衡大30 倍的热电压。Fu 等[119]采用分子动力学模拟方法研究了被限制在带电壁之间的氯化钠和氯化碘溶液的热电响应。结果表明计算得到的热电响应比标准图方法的预测大2个数量级,这表明纳米流体系统的集热性能可与最佳热电材料的集热性能相媲美。这是因为水的过剩焓(在标准图方法中被忽略)与液固界面的电渗透迁移率结合起主导作用。因此,使用水动力滑移较大的表面可以提高热电响应。
4 用于电化学相关材料设计的高通量计算和机器学习
4.1 高通量计算和机器学习概述
研发清洁高效且廉价的电化学相关材料对当前世界能源危机的解决具有重大意义,但一般从产品研发到投入市场的时间跨度极大。传统的发现新材料的方法,如经验试错法和基于密度泛函理论(DFT)的方法[120-122],由于其开发周期长、效率低、成本高,已经无法跟上当今材料科学的发展步伐。用于高通量筛选的现代化学模拟工具包可以帮助人们加速寻找新材料。特别是,通过高通量计算,无需实验合成就可以得到材料的一些关键特性[123-124]。而结合数据挖掘技术,以机器学习为核心算法的人工智能技术由于其计算成本低、开发周期短,加上强大的数据处理能力和高预测性能,正在材料检测、材料分析和材料设计中广泛应用[125]。
因此,本节主要讨论了高通量计算和机器学习在电化学相关材料的性能预测和稳定性评价、构效关系的建立等方面的成功应用。
(1)高通量计算 高通量计算通过将组合化学中的“构建单元”和“高通量筛选”观点用于材料模拟中,通过材料计算寻找或筛选材料组成的基本构建单元,构造新的化合物,建立起材料成分、结构、和性能的定量关系模型来用于指导新材料设计。
(2)机器学习 机器学习作为人工智能的一个分支,在算法的指导下,利用大量的数据不断优化模型,作出合理的预测。图12 显示了一个完整的机器学习过程,包括数据处理、建模和验证[126]。

图12 机器学习的流程(包括数据处理、建模和验证)[126]
4.2 材料性能预测和稳定性评价
目前,用于锂离子电池的电极材料种类繁多,其实际使用寿命也受到多种因素影响,难以预测。因此,如何准确预测锂离子电池等复杂非线性系统的寿命对于加速技术发展至关重要。然而,不同的老化机制、显著的设备变异性和动态的工作条件仍然是主要的挑战。现有的预测锂离子电池剩余寿命的方法大多依赖于对老化机理的先验知识,使用化学或物理配方和分析电池模型。这种依赖性在实践中通常很难确定,这限制了这些方法的应用。Severson 等[127]生成了一个包括124 个在快速充电条件下循环的商业磷酸铁锂/石墨电池的数据集,其循环寿命变化很大,从150次到2300次不等。利用早期循环的放电电压曲线(尚未显示容量退化),应用机器学习工具根据循环寿命预测和分类电池。模型在使用前100个周期(显示从初始容量中位数增加0.2%)定量预测周期寿命时达到9.1%的测试误差,使用前5个周期将周期寿命分为两组时达到4.9%的测试误差。Yang 等[128]提出了一种基于统计知识的锂离子电池健康状态估计方法。采用扩展卡尔曼滤波-递归-最小二乘联合算法对电池充电状态进行估计,同时识别模型参数和开路电压。然后采用粒子群优化-最小二乘支持向量回归方法,给出了可靠的健康状态估计结果,具有较高的精度和良好的泛化能力,粒子群优化算法提高了算法的全局寻优能力。
超级电容器作为一种清洁的储能装置,具有快速充放电动力学、高功率密度和长循环寿命等显著优点,已被广泛应用于为汽车电动机提供动力。精确评估超级电容的剩余使用寿命,为更换损坏电池提供反馈,以维持电动汽车的安全性。目前基于数据或模型的预测的评估方法要么耗时,要么精度较低。Zhou 等[129]提出了一种基于递归神经网络方法和混合遗传算法相结合的通用策略,以实现对剩余使用寿命的有效和鲁棒性评估。序列二次规划作为遗传算法的局部搜索算子,增强了遗传算法的全局搜索能力,以退出概率和隐含层单元数为手段快速搜索局部最优解。最后,将这种预测方法应用于稳态充电模式下的超级电容器,成功地估算了其剩余使用寿命。
虽然已经有大量关于溶剂对超级电容器性能的影响的实验和模拟研究,但它们往往耗时或缺少微观机理的认识。因此,需要一种更全面的方法来更好地理解溶剂在超级电容器上的作用。Su 等[130]采用将机器学习、实验和分子建模相结合的方法,研究溶剂对电容性能的影响,并可用于预测和提高超级电容器的性能。用已发表的实验数据测试了5种机器学习模型,包括支持向量回归、多层感知、M5 模型树、M5 规则和线性回归[图13(a)],确定了每个溶剂变量对电容的相对重要性。溶剂分子种类和介电常数是超级电容器最重要的变量,而溶剂的偶极矩、黏度和沸腾温度对其影响不大。Jain等[131]将人工神经网络技术应用于碳基超级电容器的电容预测。从数百篇发表的论文中提取数据训练神经网络模型,选取了比表面积、孔径、ID/IG比、氮掺杂水平和电压窗5 个特征来计算它们对电容的影响,然后选取几个碳基样本对神经网络的性能进行评价。结果表明,与线性回归、Lasso 回归等其他机器学习方法相比,人工神经网络技术在电容预测方面表现出最好的准确性和适应性。

图13 材料性能预测和稳定性评价示意图[130,132]
Yan等[132]提出了一种磷酸盐作为正极材料的高通量从头算分析方法。容量、电压、比能量、能量密度和热稳定性的评估计算数以千计的化合物[图13(b)]。确定了磷酸盐化学固有的重量和容量方面的极限,给出了磷酸盐中所有氧化还原偶的电压范围,分析了影响电压的结构因素。重新研究磷酸盐材料是否本质上是安全的,并发现在相同的氧化态下,磷酸盐的热力学释氧温度低于氧化物的。Joshi 等[133]使用深度神经网络、支持向量机和核脊回归,结合化合物的属性和其成分的元素属性的特征向量,来预测金属离子电池电极材料的电压。首先利用它们生成电压剖面图,并将其与密度泛函理论计算进行比较,最后利用该方法提出了近5000 种钠离子和钾离子电池的候选电极材料。
4.3 构效关系的建立
随着高压可充电电池等储能系统的发展,高效地寻找新的电解质变得至关重要。Park等[134]建立了一个1000000氧化还原势组成的搜索地图,以筛选具有新的核心和官能团的有机添加剂和溶剂,从而建立电解质数据库。阐明了氧化还原势与官能团之间的定量关系,最后筛选了一种喹诺林化合物作为阳极添加剂,并将其应用于电解质中,实验结果表明,在锂离子电池循环近200次时,容量保持率从64.3%提高到80.8%。高电压和高热安全性是正极材料的理想特性,但很难同时实现。Jain等[135]使用高通量密度泛函理论计算来评估超过1400 种正极材料的电压和安全性(由热力学氧气释放温度估计)之间的联系。结果表明电压与安全性之间存在强烈的反比关系。并研究了聚阴离子基团、氧化还原偶联和氧与反阳离子比对电压和安全性的影响。总的来说,只有少数化合物代表有限的氧化还原偶/聚阴离子组合,表现出高电压和高安全性。
5 结语
在目前电化学领域的高速发展中,化工热力学相关技术和应用对其发展具有很大的指导意义。在经典热力学的基础上,各种新兴的热力学领域与研究方法如雨后春笋般蓬勃发展,对推动电化学行业进步起到了辅助与加速作用。在经典化工热力学、分子和统计热力学的坚实基础上,非平衡态热力学的发展越来越贴近实际,可以定量解决大量电化学过程的模拟计算问题。高通量计算和机器学习则是近年来研究人员所使用的新方法,是用于电化学相关材料设计的利器,也是一个未来可以大有作为的化工热力学分支。如何构建完备的非平衡态热力学体系,如何高效利用新兴计算机技术来辅助热力学的发展,应当是今后发展中应重点思考和解决的问题。目前,对于实际电化学问题的解决缺乏连贯性与系统性,更加偏向于逐个攻破,问题的解决应该着力于进行多尺度多物理场的耦合计算,建立起电化学领域的理论计算体系,可以从电子尺度、原子尺度、分子尺度、颗粒尺度、器件尺度等多尺度进行连贯的计算,并且耦合电场、速度场、温度场和反应等多物理场的效应。在热力学的多重研究的加持下,电化学领域将持续高速发展,在解决人类能源问题中占有越来越重要的地位。
猜你喜欢
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01
陶瓷学报(2021年5期)2021-11-22
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
化工管理(2021年7期)2021-05-13
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2021年1期)2021-04-13
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
中学生理科应试(2019年3期)2019-07-08
