
锇炔基氢化物反应性研究
2021-09-23 08:27:36白桐桐,王铜道
大连理工大学学报 2021年5期
白 桐 桐,王 铜 道
(大连理工大学 张大煜(化学)学院,辽宁 大连 116024)
0 引 言
过渡金属炔基配合物由于其多元化的分子构型、丰富的光致发光性能引发了巨大的研究热潮,特别是作为一种新型的有机发光材料,在现代科技领域发挥着重要的作用[1-2].炔基单元的线性构型和π共轭性使其与金属中心存在高度灵活的键合模式,如形成σ共价键或dπ-pπ相互作用[3-4].金属氢化物在金属有机化学中扮演着重要的角色,因其可以与很多不饱和有机化合物发生插入反应生成含M-C键的反应中间体,因而备受关注.鉴于很多化学反应的关键步骤是金属氢化物的插入[5],因此对于金属氢化物的研究不仅在合成化学方面意义重大,而且对于催化领域也意义非凡.金属炔基氢化物兼具金属炔基配合物和金属氢化物的结构属性,受到了广大化学工作者的广泛关注,但是目前文献上关于其合成方法和反应性的研究并不多.这主要是由于其结构大都不稳定,因此其相关的化学研究受到了极大的限制.
金属炔基氢化物的合成方法主要有下列3种:(1)通过末端炔与不饱和过渡金属配合物氧化加成[6];(2)通过炔基阴离子取代过渡金属配合物卤素等配体[7];(3)通过二氢金属配合物与末端炔烃的反应[8].金属炔基氢化物由于其特定的碳碳三键和金属氢结构特性,可以发生配体取代反应[8]、分子内重排反应[9]及其他一些有趣的金属有机反应[1,10-13].其中分子内重排反应是金属炔基氢化物最常见的反应之一,比如金属炔基氢化物发生氢转移生成金属亚乙烯基配合物.目前对于金属炔基氢化物的合成及反应性研究主要集中于铑、钌、铱等过渡金属上.总体而言,金属炔基氢化物的反应性研究仍然比较少[14-15].
鉴于上述情况,本文设计由锇炔基氢化物OsH(CO)(C≡CTMS)(PPh3)3(1)合成稳定的OsH(CO)2(C≡CTMS)(PPh3)2(2),并进一步探索锇炔基氢化物的化学反应性,比如配合物2与三乙胺盐酸盐的反应,以及配合物1与不同化学计量的[Cp2Fe]+[BF4]-/H2O的反应,通过核磁共振谱、高分辨质谱、X射线单晶衍射对所有产物进行表征.
1 实验部分
1.1 仪器与试剂
仪器:核磁共振谱在Bruker AV400 (400 MHz)核磁共振仪上测定;X射线单晶衍射在Bruker Smart APEX Ⅱ上测定,若无特别说明,操作温度为200 K;高分辨质谱均在Agilent 6224 TOF LC/MS上测定.
试剂:本文所涉及的原料均购于安耐吉、阿拉丁等正规渠道,若无特殊说明,实验均在氮气保护下的Schlenk反应瓶中进行.二氯甲烷、四氢呋喃、正己烷等均为经溶剂纯化系统处理过的无水无氧溶剂.
1.2 实验方法
配合物2在干燥洁净的100 mL厚壁耐压瓶中,称取配合物1(1 101.2 mg,1.00 mmol),加入20 mL二氯甲烷,得到无色溶液.然后将此耐压瓶置于-30 ℃条件下,真空除去耐压瓶中的气体,在室温下将8 Pa的一氧化碳气体通入.在60 ℃ 条件下,混合液反应3 d,真空除去溶剂及残余的一氧化碳气体,将所得固体用正己烷(10 mL)洗涤3次,过滤并真空干燥,可得到纯白色固体产物2.产量为783.6 mg,产率为90%.1H NMR(400 MHz,299 K,CDCl3)δ:7.34~7.81(m,45H,Ph),-0.39(s,9H,Si(CH3)3),-6.24(t,2J(PH)=20.8 Hz,1H,OsH).31P{1H}NMR(162 MHz,299 K,CDCl3)δ:6.46(s,PPh3).1H,13C GHSQC(400 MHz/101 MHz,299 K,CDCl3)δ1H/δ13C:-0.39/0.61(Si(CH3)3).1H,13C GHMBC(400 MHz/101 MHz,299 K,CDCl3):δ1H/δ13C:-0.39/118.88(Si(CH3)3/CCSi(CH3)3),-6.24/116.45,182.11(OsH/CCSi(CH3)3,Os(CO)2).13C{1H}NMR(101 MHz,299 K,CDCl3)δ:182.48,181.49(t,2J(PC)=6.1 Hz,Os(CO)2),127.81~135.34(Ph),118.88(t,3J(PC)=2.0 Hz,CSi(CH3)3),116.45(t,2J(PC)=17.2 Hz,CCSi(CH3)3),0.61(Si(CH3)3).HRMS(ESI):893.178 0;[M+Na]+(C43H40O2SiP2OsNa理论值:893.178 0).
配合物3在干燥洁净的100 mL厚壁耐压瓶中,称取配合物2(300.2 mg,0.35 mmol)和三乙胺盐酸盐(50.4 mg,0.37 mmol),加入20 mL二氯甲烷和1 mL甲醇,在60 ℃条件下混合液反应约3 d.将所得产物抽干,以二氯甲烷和正己烷体积比为1∶1作为洗脱剂进行柱色谱(氧化铝)分离,收集组分并抽干后得到白色固体3.产量为279.3 mg,产率为89%.1H NMR(400 MHz,299 K,CDCl3)δ:7.98(d,3J(HH)=17.1 Hz,1H,CH=CHSi(CH3)3),7.34~7.65(m,30H,Ph),5.83(d,3J(HH)=17.2 Hz,1H,CH=CHSi(CH3)3),-0.46(s,9H,Si(CH3)3),31P{1H}NMR(162 MHz,299 K,CDCl3)δ:-8.83(s,PPh3).HRMS(ESI):929.151 2;[M+Na]+(C43H41ClO2SiP2OsNa 理论值:929.154 7).
配合物4在干燥洁净的100 mL Schlenk反应瓶中,称量配合物1(205.6 mg,0.19 mmol)和[Cp2Fe]+[BF4]-(127.0 mg,0.47 mmol),加入15 mL二氯甲烷,充分搅拌,室温反应约13 h后,得到深绿色悬浊液.用低孔径滤膜进行过滤得到深绿色溶液,除去溶剂,将所得固体溶于二氯甲烷(5 mL)中,再加入正己烷(50 mL)后析出翠绿色的固体,过滤并干燥后得到翠绿色的固体产物4.其产量为159.0 mg,产率为77%.1H NMR(400 MHz,299 K,CDCl3)δ:6.84~7.81(m,45H,Ph),2.92(s,2H,OsOH2),-6.99(dt,2J(PH)=84.3,25.1 Hz,1H,OsH).31P{1H}NMR(162 MHz,299 K,CDCl3)δ:15.12(d,2J(PP)=11.1 Hz,Os(P1(3)Ph3)2),2.81(t,2J(PP)=10.9 Hz,OsP2Ph3).
配合物5在干燥洁净的100 mL Schlenk反应瓶中,称量配合物1(223.7 mg,0.20 mmol)和[Cp2Fe]+[BF4]-(68.0 mg,0.25 mmol),加入15 mL二氯甲烷,充分搅拌,室温反应约15 h后,得到墨绿色悬浊液.用低孔径滤膜进行过滤得到墨绿色溶液,除去溶剂,将所得固体溶于二氯甲烷(5 mL)中,再加入正己烷(50 mL)后析出浅绿色固体,过滤并干燥后得到浅绿色固体产物5.产量为166.3 mg,产率为73%.1H NMR(400 MHz,299 K,CDCl3)δ:5.83~7.84(m,44H,Ph),4.42(t,J(HH)=9.1 Hz,1H,OsCH),2.57,2.40(s,1H,OsCH2),-4.58(dt,2J(PH)=25.1,12.3 Hz,1H,OsH).31P{1H}NMR(162 MHz,299 K,CDCl3)δ:11.25(t,2J(PP)=22.2 Hz,1P,OsPPh2),8.33(dd,2J(PP)=140.9,22.5 Hz,1P,OsPPh3),-5.73(dd,2J(PP)=140.9,22.1 Hz,1P,OsPPh3).1H,1H GCOSY(400 MHz/400 MHz,299 K,CDCl3)δ1H/δ1H:4.42/2.57,2.40(OsCH/OsCH2,OsCH2),1H,13C GHSQC(400 MHz/101 MHz,299 K,CDCl3)δ1H/δ13C:4.42/72.01(OsCH),(2.57,2.48)/49.76(OsCH2).1H,13C GHMBC(400 MHz/101 MHz,299 K,CDCl3):δ1H/δ13C:4.42/49.76(OsCH/OsCH2),2.57/72.01(OsCH2/OsCH),-4.58/49.76,72.01,178.87(OsH/OsCH2,OsCH,CO).13C{1H}NMR(101 MHz,299 K,CDCl3)δ:178.87(CO),122.64~152.26(Ph),72.01(OsCH),49.76(OsCH2).HRMS(ESI):1 033.253 0;[M-BF4]+(C57H48OP3Os理论值:1 033.252 7).
2 结果与讨论
2.1 锇炔基氢化物的配体取代反应
由于反位效应,配合物1中金属氢原子可以弱化处于其反位的Os-PPh3键,使Os-PPh3键变长,此外三苯基膦基团具有很大的位阻效应,从而使得该三苯基膦基团更容易被取代.基于此种思考,探索了配合物1与一氧化碳的反应性.以厚壁耐压瓶为反应容器,将配合物1溶于少量二氯甲烷后低温下抽至负压,然后通入一氧化碳气体,在60 ℃条件下反应3 d后,真空除去溶剂及气体,经洗涤,以90%的产率得到配合物2(图1).该配合物中的一氧化碳取代锇氢原子对位三苯基膦基团,其稳定性比配合物1大大提高.其结构得到了X射线单晶衍射、核磁共振谱以及高分辨质谱的确认.
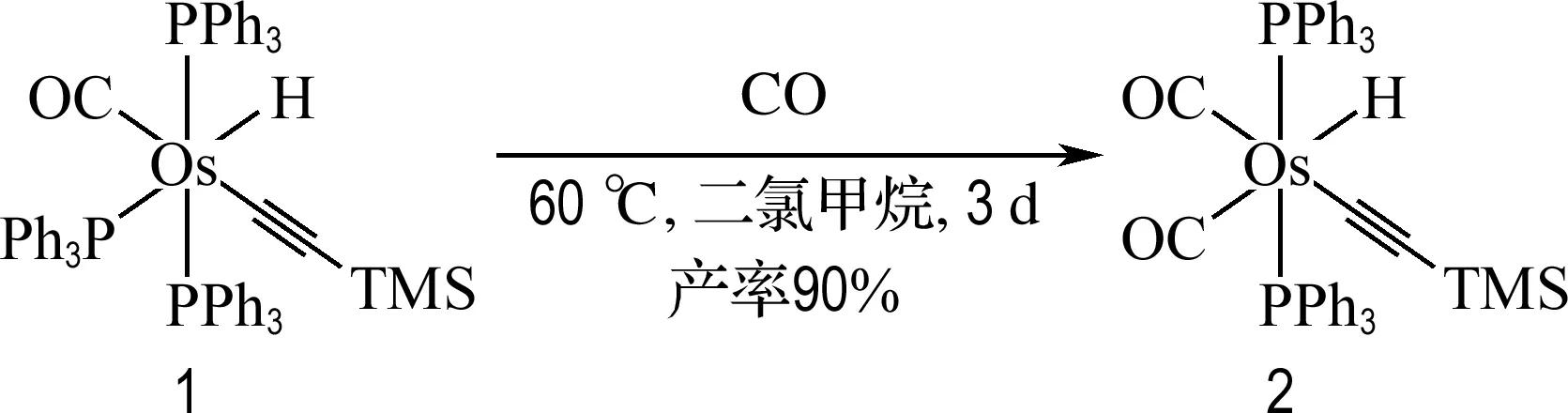
图1 配合物2的合成Fig.1 Synthesis of complex 2
在1H NMR谱图中,δ=-6.24处存在一个明显的三重峰,为邻位两个三苯基膦基团和对位一个三苯基膦基团对锇氢的裂分[16],偶合常数2J(PH)=20.8 Hz.-0.39处的化学位移为三甲基硅基中的甲基氢信号,其与锇氢原子的峰面积积分比例约为9∶1.在δ=7.34~7.81处,可观察到苯环上氢的多重峰信号.在31P{1H}NMR谱图中,化学位移为6.46处存在一个单峰,此为与锇原子相连的两个化学环境等价三苯基膦基团的磷信号.
此外,对配合物2进行了高分辨质谱测试,其理论分子式为C43H40O2SiP2Os,理论相对分子质量为870.188 8.在ESI作为电离源,以二氯甲烷和乙腈体积比为1∶1作为溶剂的条件下,观察到配合物2的加钠离子峰[配合物2+Na]+:893.178 0,其理论出峰位置为893.178 0,误差为0.
对配合物2进行单晶培养并得到无色块状晶体(两相法:将产物溶于二氯甲烷,然后覆盖正己烷,-25 ℃下静置2 d).X射线单晶衍射结构(图2)显示一氧化碳取代氢原子对位的三苯基膦基团.配合物2可以看作一个扭曲的八面体结构,两个三苯基膦基团处于锇原子轴向位置,而C1、O1、C2、O2、C3、C4、Si1占据锇原子的赤道平面位置.配合物2的C3-C4的键长为0.120 6(4)nm,这是典型的碳碳三键键长.Os1-C3-C4的键角为178.6(3)°,P1-Os1-P2的键角为170.3(2)°.

图2 配合物2的X射线单晶衍射结构(CCDC-2058302)Fig.2 Single-crystal X-ray diffraction structure of complex 2 (CCDC-2058302)
2.2 配合物2与三乙胺盐酸盐的反应
配合物2与三乙胺盐酸盐混合物在60 ℃条件下反应3 d后,经中性氧化铝柱色谱分离,可得到锇炔基还原为锇乙烯基产物3(图3),产率为89%.其结构也得到了X射线单晶衍射、核磁共振谱以及高分辨质谱的确认.

图3 配合物3的合成Fig.3 Synthesis of complex 3

此外,对配合物3进行了高分辨质谱测试,其理论分子式为C43H41ClO2SiP2Os,理论相对分子质量为906.165 4.在ESI作为电离源,以二氯甲烷和乙腈体积比为1∶1作为溶剂的条件下,观察到配合物3的加钠离子峰[配合物3+Na]+:929.151 2,其理论出峰位置为929.154 7,相对误差为3.8×10-6.
如图4所示,配合物3也是一个扭曲的八面体结构,锇氢原子被氯原子取代,同时炔基还原为反式烯基,两个三苯基膦基团处于锇原子轴向位置.C1、O1、C2、O2、C3、C4、Si1、Cl1占据锇原子的赤道平面位置.C3-C4的键长为0.134 3(11)nm,这是典型的碳碳双键键长.反式烯基中Os1-C3-C4的键角为130.2(6)°,C3-C4-Si1的键角为126.3(7)°,这些均为典型碳碳双键键角.

图4 配合物3的X射线单晶衍射结构(CCDC-2058303)Fig.4 Single-crystal X-ray diffraction structure of complex 3 (CCDC-2058303)
如图5所示,推测了锇乙烯基配合物3的生成机理.首先锇炔基的β-碳具有亲核性,进攻三乙胺盐酸盐中H+,得到中间体3A;然后中心金属锇上氢原子发生1,2-氢迁移得到中间体3B结构;最后游离的Cl-配位到锇中心的空位上,得到18e稳定结构,即配合物3.
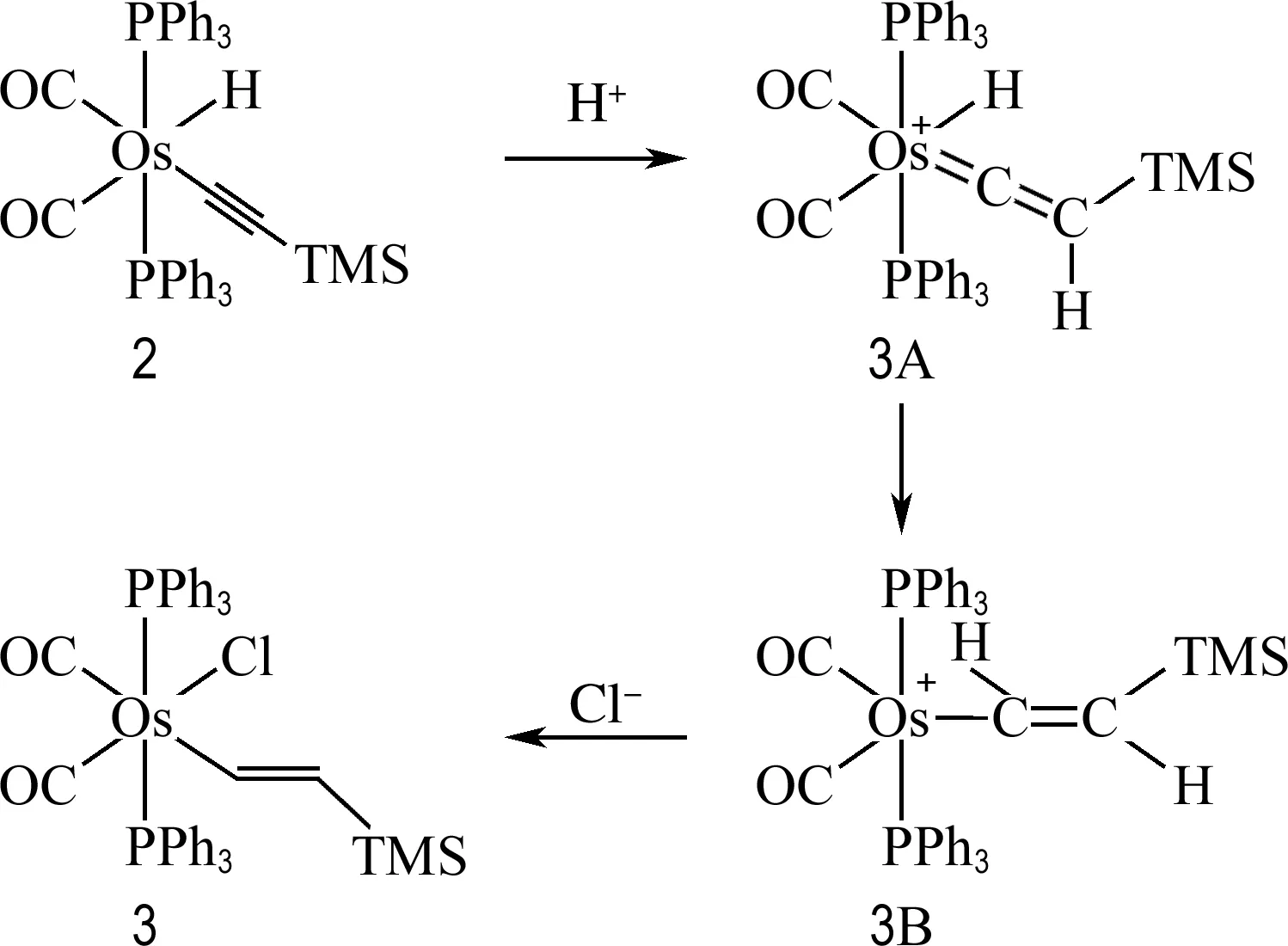
图5 配合物3可能的生成机理Fig.5 Possible mechanism for the formation of complex 3
2.3 配合物1与[Cp2Fe]+[BF4]-/H2O的反应
[Cp2Fe]+[BF4]-是一种被广泛应用的单电子氧化剂[17-18].在探索配合物1与其反应性时,根据化学计量比的差异,得到了两种完全不同的产物4和5(图6).配合物1与2.5当量的[Cp2Fe]+[BF4]-/H2O室温下可发生反应,得到单一的翠绿色固体产物4,产率为77%;当配合物1和[Cp2Fe]+[BF4]-/H2O的反应摩尔比为1∶1时,在室温条件下反应15 h,得到浅绿色固体产物5,产率为73%.X射线单晶衍射、核磁共振谱和高分辨质谱确定了配合物4和5的结构.
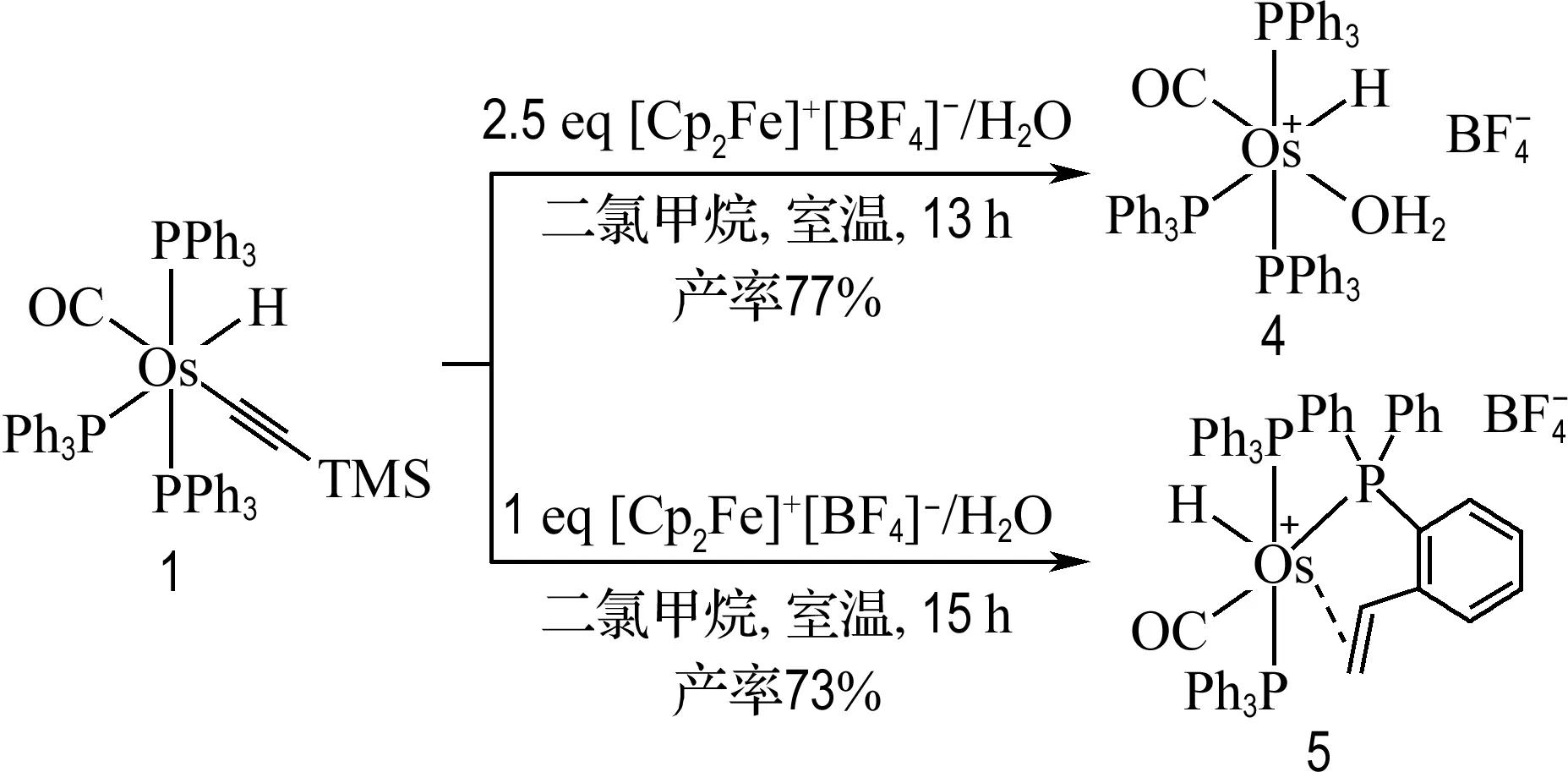
图6 配合物1与[Cp2Fe]+[BF4]- /H2O的反应Fig.6 Reaction of complex 1 with [Cp2Fe]+[BF4]-/H2O
在配合物4的1H NMR谱图中,δ=2.92处的单峰归属为OsOH2的氢信号.OsH的氢信号在δ=-6.99处,为dt裂分峰.在31P{1H}NMR谱图中,δ=15.12处的两重峰为轴向两个化学环境等价三苯基膦基团的磷信号,δ=2.81处的三重峰为处于赤道平面的三苯基膦基团的磷信号.
在配合物5的1H NMR谱图中,OsH的信号在δ=-4.58处,峰型为dt.OsCH的信号在δ=4.42 处.δ=2.57和2.40处的信号归属于OsCH2,为两重峰,其与OsCH信号的峰面积积分比例为2∶1.在31P{1H} NMR谱图中,δ=11.25 处的三重峰为含有被活化苯环三苯基膦基团的磷信号,δ=8.33和-5.73处的两个dd裂分峰为锇原子轴向两个三苯基膦基团上的磷信号.


图7 配合物4的X射线单晶衍射结构(CCDC-2058304)Fig.7 Single-crystal X-ray diffraction structure of complex 4 (CCDC-2058304)
图8列出了配合物4可能的生成机理.首先配合物1与[Cp2Fe]+[BF4]-反应,锇中心由+2价被氧化成+3价得到中间体4A;进一步地,[Cp2Fe]+[BF4]-氧化锇中心至+4价,得到中间体4B;最后锇中心发生还原消除,离去TMS—C≡C+和BF4-组分(可能进一步反应为TMS—C≡C—F和BF3,实验中未对副产物进行表征和分离),同时H2O中的O配位至锇中心,得到稳定产物4.
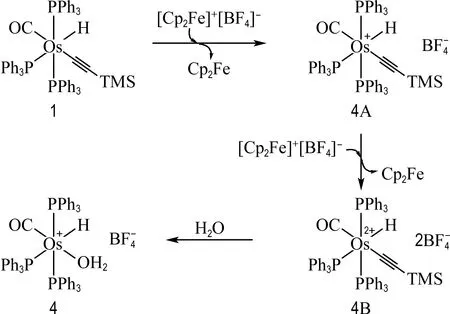
图8 配合物4可能的生成机理Fig.8 Possible mechanism for the formation of complex 4
在配合物5的X射线单晶衍射结构(图9)中,炔基被还原为烯基并且与赤道平面上的一个三苯基膦基团的苯环邻位碳发生偶联.锇原子轴向位置的两个三苯基膦基团由于受到赤道平面上的巨大空间位阻作用,两个三苯基膦基团发生较大的偏离,P1-Os1-P3键角为146.80(10)°,比配合物4小约13°,偏离180°约33°.C2-C3的键长为0.141 4(14)nm,介于碳碳单键和碳碳双键之间,这是由于锇金属中心与C2-C3双键的离域π键作用使得其键长被拉长.

图9 配合物5的X射线单晶衍射结构(CCDC-2058305)Fig.9 Single-crystal X-ray diffraction structure of complex 5 (CCDC-2058305)
如图10所示,推测了配合物5的生成机理.首先[Cp2Fe]+[BF4]-和H2O原位形成的HBF4中的质子受到炔基β-碳的亲核进攻,生成金属亚乙烯阳离子中间体5A;随后TMS基团水解得到中间体5B,5B发生1,2-氢迁移生成5C;接着赤道平面上三苯基膦基团的苯基邻位C-H对中心金属锇氧化加成生成中间体5D;经过构型翻转得到5E,最后发生还原消除反应得到乙烯基配位到锇中心的结构5,其共振结构式为5′.

图10 配合物5可能的生成机理Fig.10 Possible mechanism for the formation of complex 5
3 结 语
本文通过对锇炔基氢化物OsH(CO)(C≡CTMS)(PPh3)3(1)结构特征的分析,经配体取代反应,高产率地得到了更稳定且含有两个羰基配体的锇炔基氢化物OsH(CO)2(C≡CTMS)(PPh3)2(2).配合物2可以与三乙胺盐酸盐在加热条件下反应,其碳碳三键被还原为碳碳双键,得到锇乙烯基配合物3,该反应实现了金属炔基氢化物向金属乙烯基配合物的转化.此外,配合物1与不同计量比的[Cp2Fe]+[BF4]-/H2O的反应,经过复杂的转化过程,可分别单一地得到配合物4和5.所有产物都得到了核磁共振谱、高分辨质谱以及X射线单晶衍射的表征.上述研究结果进一步丰富了金属炔基氢化物的反应性.
猜你喜欢
山东冶金(2022年4期)2022-09-14 08:59:08
经济技术协作信息(2018年11期)2019-01-14 03:07:22
上海化工(2018年10期)2018-10-31 01:21:06
环境保护与循环经济(2017年12期)2017-07-11 01:46:36
中国科技信息(2016年19期)2016-10-25 08:15:06
中国科技信息(2016年6期)2016-08-31 07:27:16
中国科技信息(2015年24期)2015-11-07 08:52:23
中国科技信息(2015年23期)2015-11-07 08:25:56
化工生产与技术(2014年5期)2014-02-27 13:42:02
化学分析计量(2013年4期)2013-03-11 16:37:37
