
基于片段组装的蛋白质结构预测方法综述
2021-09-20 10:25张贵军赵凯龙
数据采集与处理 2021年4期
张贵军,刘 俊,赵凯龙
(浙江工业大学信息工程学院,杭州 310012)
引 言
蛋白质是生命活动的主要承担者,几乎支撑着生命的所有功能,细胞内发生的大部分反应都依赖于蛋白质。蛋白质的功能取决于其独特的三维结构,也就是常说的“结构决定功能”。随着2003年人类基因组计划宣布完成[1],由DNA 或RNA 转译为蛋白质氨基酸序列的第一遗传密码已被破解,然而蛋白质序列折叠成特定的三维结构才能够执行其特定的功能。蛋白质序列如何折叠形成独特的三维结构仍然是未解之谜[2]。《Science》杂志在纪念创刊125 周年之际,把“能否预测蛋白质折叠?”列为21 世纪125 个科学前沿问题之一[3]。因此,对蛋白质折叠过程的深入研究,对于直接、准确地分析蛋白质的生物学功能和解释各种生命活动现象至关重要,将为相关疾病的诊断与治疗、创新药物研发奠定基础。
目前,主要通过X 射线衍射、核磁共振和冷冻电镜等生物实验手段来测定蛋白质的三维结构,这些方法不仅费钱费力,而且周期长,导致已测定蛋白质结构的数量远远低于已测定蛋白质的序列数量。2021年4 月最新统计数据显示,UniProtKB/TrEMBL 数据库中共存储蛋白质序列214 406 399 条(数据来源于http://www.ebi.ac.uk/uniprot/TrEMBLstats),其中177 426 条序列结构被实验测定(数据来源于http://www1.rcsb.org/stats/growth/growth‑released‑structures),仅占序列总数的0.083%,而且这一差距仍然在不断增加。显然,实验测定方法无法满足高效获取蛋白质结构的需求。
在理论研究和实际应用双重需求的推动下,依据Anfinsen 准则[4],通过计算机技术根据氨基酸序列预测三维结构的蛋白质结构预测取得了蓬勃发展。CASP 竞赛是由美国科学家Moult 发起的蛋白质结构预测技术关键评估活动,能够客观地反映蛋白质结构预测领域发展的最新技术水平,是蛋白质结构预测领域的奥林匹克竞赛[5]。CASP 竞赛每两年举行一次,自1994年创办至今已举办14 届。CASP 根据目标蛋白预测难易程度分为基于模板(Template‑based modeling,TBM)和无模板(Free modeling,FM)两类建模方法[6]。一般来讲,TBM 方法中目标蛋白可以从PDB(Protein data bank)结构数据库中检测到同源模板,建模精度基本能够达到实验测定水平[5];然而,由于无法获得同源模板,FM 类目标蛋白必须采用从头预测方法,成为CASP 中最具挑战、也是最受关注的一类研究问题。能量模型的复杂性和构象空间采样瓶颈是限制从头预测方法发展的主要原因[7]。
从头蛋白质结构预测不受限于模板信息,能够正确预测具有未发现的整体拓扑结构的蛋白质结构,一直受到生物信息学领域和进化计算社区的高度关注。片段组装技术在从头蛋白质结构预测领域应用广泛,事实证明片段组装方法是最有前景的蛋白质结构预测方法之一[6,8]。本文结合国内外研究现状以及本课题组开展的一些研究工作,针对基于片段组装的蛋白质结构从头预测方法的研究进展进行分析和综述。
1 片段组装
由于蛋白质构象空间的高维特性,在巨大的构象空间中进行采样是不合适的。片段组装技术利用已测定蛋白质结构的局部信息,将每一个残基的二面角约束在一组离散值内,从而极大地缩小了构象搜索空间[9‑10]。在蛋白质结构预测中,片段组装技术分为3 个步骤:首先,随机在目标序列上选择一个包含若干个(一般为3 个或9 个)连续残基的插入窗口;然后,从该窗口对应的片段库中随机选择一个片段替换该窗口对应的片段;最后,采用能量函数计算片段替换前后构象的能量差值,并根据Metropolis 准则判断是否保留片段组装后的构象[9]。图1 为长度为L的蛋白质进行3 残基片段组装的示意图。片段组装利用PDB 数据库中已测定蛋白质结构的短且连续的片段信息,在基于知识力场构建的能量函数的引导下,不断组合向天然态折叠,既利用了已知蛋白质的结构信息,同时避免了同源建模方法高度依赖模板质量的缺陷[11]。
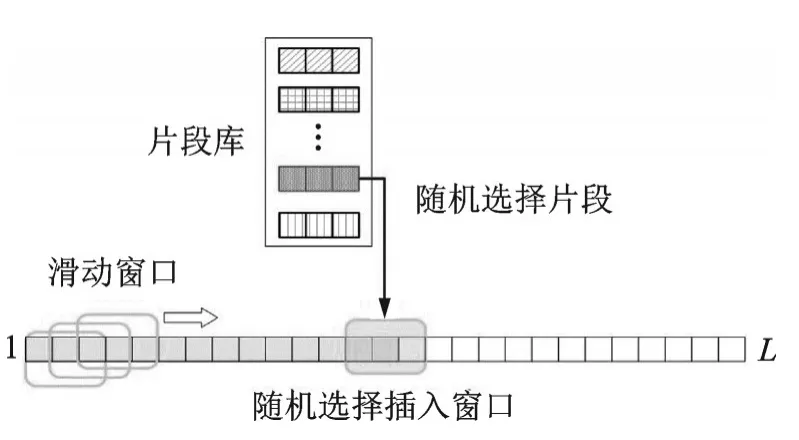
图1 片段组装示意图Fig.1 Schematic diagram of fragment assembly
2 国内外研究现状
蛋白质结构预测一直受到计算生物学领域和计算智能社区的高度关注,是一个前沿研究课题[12]。1994年,Bowie 和Eisenberg 首次从PDB 中提取序列长度为9 的小片段来组装形成一个新的三维结构[13]。此后的二十多年间,片段组装成为广泛使用的从头蛋白质结构预测方法。虽然自从2016年CASP12 深度学习在蛋白质残基接触/距离预测取得重大突破后,基于距离约束的几何优化方法逐渐占据主导地位[14‑15],但从CASP12 至CASP14(2016—2020年)的结果中可以发现片段组装方法仍然是最具竞争力的从头蛋白质结构预测方法之一[8,16]。国内外研究学者针对基于片段组装的结构预测做了大量深入研究[17‑18],本文将从经典的片段组装结构预测方法、基于进化算法的片段组装方法和残基接触距离辅助的片段组装方法这3 方面进行介绍。
2.1 经典片段组装方法
华盛顿大学Baker 实验室开发的Rosetta[11,19]是较早采用片段组装技术的从头蛋白质结构预测方法。在Rosetta 中,已知结构的短片段通过蒙特卡洛策略组装,以产生类似天然的蛋白质构象。Rosetta通过能量力场来描绘蛋白质折叠过程中不同状态的构象,根据热力学假说,天然态的蛋白质结构对应于自由能最低的构象,通过最小化构象能量获取近天然态构象。由于蛋白质构象空间极其复杂,为了提高采样效率,通常采用Rosetta 低分辨率能量函数score3 来减小自由度,同时保留重要信息。能量函数score3 由10 个能量项组成,反映原子排斥、氨基酸倾向、残基环境、残基对相互作用、二级结构元素之间的相互作用、密度和紧致性等,其定义为[11]
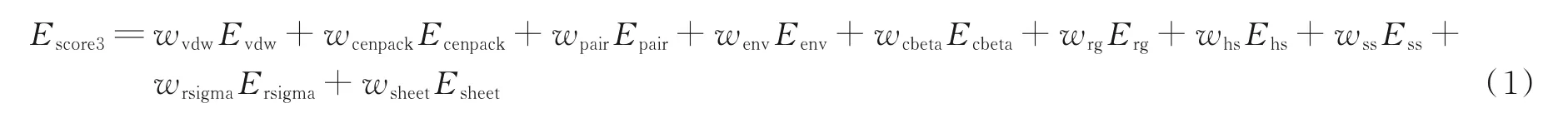
Rosetta 片段组装折叠模拟主要分为4 个阶段,在每个阶段采用不同的能量函数,每个能量项的权重逐渐增加。在Rosetta 的前3 个阶段使用残基数目为9 的片段执行片段组装,实现大规模的构象空间探索,在第4 阶段使用残基数目为3 的片段来更精细地调整构象拓扑结构。Rosetta 的每个阶段执行大量的片段插入,并根据片段插入情况动态调整温度因子。当片段插入连续失败150 次,通过提高温度因子来降低构象接受的条件,从而提高片段插入成功率;当片段插入成功后,将温度因子恢复为初始值。为了生成可靠的蛋白质模型,通常需要运行成千上万次的片段组装折叠模拟最终生成最低能量模型,这是一个极其耗时且消耗计算代价的过程。
密西根大学张阳实验室开发的QUARK[20‑21]是另一个优秀的基于片段组装的从头蛋白质结构预测方法。QUARK 使用的片段长度为1 至20 个残基,采用基于知识的复合力场引导的副本交换蒙特卡洛来从片段组装全长结构模型。为了便于力场的发展和搜索引擎的设计,QUARK 采用半简化模型,用主干原子和侧链质心来表示蛋白质残基。对于查询序列,首先通过神经网络预测各种结构特征。然后通过副本交换蒙特卡洛模拟,将无缝穿线生成的小片段组装起来,从而生成全局折叠。QUARK 设计了包含11 个能量项的复合力场来引导构象搜索,总能量的计算公式为[20]
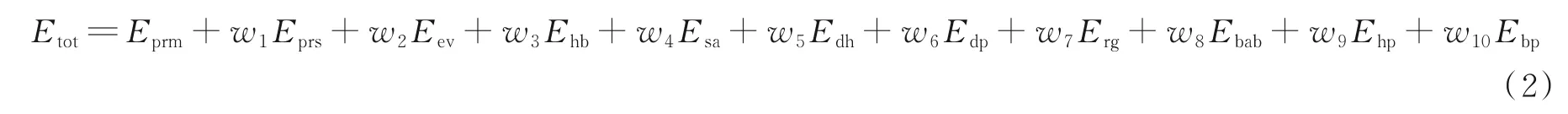
式中:Eprm、Eprs和Eev为原子级能量项,分别表示主链原子对势能、侧链中心成对势能和排除体积;Ehb、Esa、Edh和Edp为残基级能量项,分别表示氢键作用力、溶剂可及性、主链扭转角势能和基于片段的距离谱能量;Erg、Ebab、Ehp和Ebp为拓扑级的能量项,分别表示回转半径、β‑α‑β惩罚项、α‑α能量项和β对能量项。
QUARK 设计了11 个局部构象运动来增强算法的采样能力,这些局部运动分为残基级、片段级、拓扑级3 个层次,在40 个平行副本中运行蒙特卡洛模拟。虽然在低温下的模拟可以探测到较低能量的构象,但很容易陷入到局部能量盆地中。副本交换的目的是利用高温副本模拟帮助低温副本跳出局部低能源盆地。因此,对于交换每一对相邻的副本,保持高接受率是必要的。每个副本在每个周期内单独运行,其中将根据Metropolis 准则尝试30L1/2(L是蛋白质长度)次局部运动。在一个运行周期完成后,将尝试在每两个相邻副本之间进行互换操作,交换它们的诱饵构象。互换操作也遵循Metropolis 准则。与Rosetta 单纯的片段替换相比,QUARK 模拟包含了自由链结构的复合运动和结构之间的片段替换。这些技术极大地提高了构象搜索的灵活性和效率。
除Rosetta 和QUARK 之外,FRAGFOLD[22]、SCRARTCH[23]、PROFESY[24]等一系列方法都属于早期典型的基于片段组装的从头蛋白质结构预测方法。
2.2 基于进化算法的片段组装方法
进化算法[25‑26]是一种基于自然选择和遗传变异等生物进化机制的全局性搜索算法,是研究蛋白质构象优化的一类重要方法。进化算法通过交叉和变异算子以及选择策略来模拟生物进化过程,提高算法的可靠性。进化算法在蛋白质结构预测领域应用广泛,总体实现流程如下:①通过随机片段组装生成包含若干个构象的初始种群;②对种群中的父代构象进行交叉和变异操作,生成子代构象;③计算子代构象的能量,通过选择策略判断是否用子代构象替换父代构象;④迭代步骤②和③,直到满足终止条件。由于蛋白质的高维特性,能量景观中存在着大量的局部能量陷阱,蒙特卡洛算法极易陷入局部能量陷阱,使算法早熟。在进化算法的框架下通过片段组装来预测蛋白质的三维结构,无须重复运行大量独立轨迹,能够实现种群中构象的信息交互,从而提升算法的采样效率和预测精度。
Garza‑Fabre 等在Rosetta 片段组装协议的基础上提出多阶段模因算法RMA(Rosetta‑based memetic algorithm)[27]。RMA 分为4 个阶段,每个阶段都是基于标准Rosetta 片段组装的相应阶段设计的。第1 阶段,利用Rosetta 第1 阶段的随机片段组装进行种群初始化,得到一组多样化的初始构象。在第2、3 和4 阶段,首先将Rosetta 相应阶段作为局部搜索更新种群中的构象;然后将种群中的构象进行两两配对作为父代构象,利用预测的二级结构信息设计了基于loop 区域残基的重组和突变遗传算子,通过对每对父代构象执行遗传算子操作生成子代构象;最后在生存选择环节,通过同时考虑构象的能量和多样性从父代和子代构象中选择较优构象构建新的种群。日本理化学研究所的Zhang 研究小组提出了基于统计学原理的随机优化算法EDA(Estimation of distribution algorithm)的蛋白质结构预测方法EdaFold[28],通过统计构象搜索过程中的进化信息来指导当前构象搜索,以生成优秀的新构象。基于全原子力场模型,在Eda‑Fold 算法基础上,该研究小组进一步提出了EdaFoldAA方法[29]和基于聚类变异更新策略的EdaFoldC方法[17]。Baker 团队在Rosetta 基础上开发了迭代的杂化协议,整个迭代过程由进化算法指导,在每次迭代时将杂化作为变异或交叉操作,并控制构象的多样性以防止快速收敛[19],此外,根据预测的接触图与PDB中已知结构的接触图对齐以进行折叠识别,并利用接触图比对工具map_align 来挑选不连续的片段,进一步整合宏基因组数据,为614 个目前结构未知的蛋白质家族生成模型[30]。
本课题组在基于进化算法的片段组装方面进行了深入研究。由于蛋白质的高维特性,需要搜索的构象空间过于庞大,传统的片段组装方法通常分为多个阶段来搜索构象空间。针对不同蛋白质的阶段切换问题,本课题组提出了包含探索和增强两阶段的群体蛋白质结构预测算法PAIE[18],旨在通过基于熵的阶段切换策略和基于扭转角分布的选择策略来克服当前多阶段算法的局限性,确保适当搜索构象空间并进一步增强算法的探索能力。此外,根据探索阶段构象的扭转角分布设计了一种选择策略,并将其应用于增强阶段。针对能量模型的不精确性,提出了一种基于距离谱引导的差分进化算法DP‑DE[31]。在DPDE 中,设计了一种基于距离谱的选择策略来指导构象空间采样,除能量外,将残基-残基距离作为一种辅助构象评估指标,以补偿能量函数的不准确性,并基于距离分布设计了一个距离接受概率,用于选择构象。当试验构象的能量低于目标构象的能量时,试验构象直接被下一代接受。否则,首先计算从片段库中提取的残基-残基距离与从构象中所有残基对的距离分布图获得的预测残基-残基距离之间的平均距离误差。如果试验构象在平均距离误差方面优于目标构象,则计算试验构象的距离接受概率,并根据距离接受概率接受试验构象。该策略保留了具有更高能量但更合理结构的构象。通过使用基于距离谱的选择策略引导采样,提高了算法逃逸局部能量陷阱的能力和搜索效率。在距离谱研究的基础上,进一步提出了一种基于距离特征的两阶段蛋白质结构预测优化算法TDFO[32]。通过二分K‑means 算法提取距离谱中的特征信息用于构建构象相似性评估指标,并设计了基于构象相似度的选择策略引导构象采样,在一定程度上降低了不精确的能量模型的影响,同时提高了采样过程中构象的多样性;此外,根据算法的不同阶段提出了两种变异算子,并设计了一种状态估计模型以实现不同搜索阶段的平衡。
2.3 残基间接触距离辅助的片段组装方法
自CASP12 以来,基于深度学习的蛋白残基间接触(contact)预测和距离(distance)预测取得了重大进展,使得结构预测精度显著提升[5,14]。蛋白质的多序列比对蕴含着序列的进化信息,根据残基对的共变特性可以推断出它们在空间中的位置关系(是否接触或距离),研究表明远程contact 对结构预测非常有帮助,而残基间距离分布为蛋白质折叠提供了更加丰富和细粒度的约束信息。残基接触和距离预测的成功也进一步推动了基于片段组装蛋白质结构预测的发展。
早在2014年,Jones 团队就发现了将基于片段组装的折叠算法FRAGFOLD 与残基间接触预测方法PSICOV 相结合的潜在好处[33]。将PSICOV 预测的残基间接触作为能量项添加到FRAGFOLD 现有的能量函数中,并通过模拟退火将超二级结构片段和长度固定的短片段组装成三维结构。结果证明,使用残基间接触的FRAGFOLD 的预测精度得到了显著提升。在2016年的CASP12 中,张阳课题组将基于序列预测的残基间接触约束信息加入I‑TASSER 和QUARK 中,使得QUARK 在FM 目标的前5 个预测模型中最好模型的平均精度提高了37%[8];在CASP13 中,张阳团队发布了C‑I‑TASSER 和C‑QUARK,将残基间接触信息进一步优化为一个新的接触势能项,与包括基于穿线的距离约束和基于固有知识(物理势能)在内的其他能量项相平衡,以指导结构组装模拟折叠目标蛋白[16];2020年11 月召开的CASP14 会议摘要显示,基于深度学习预测的残基间距离和扭转角也被整合到I‑TASSER 和QUARK 之中,以进一步提升结构预测精度。
本课题组在片段组装的基础上,结合残基间接触距离信息,提出了一些有效的采样策略和优化方法来提升蛋白质结构预测的精度和效率。利用残基间接触和二级结构信息,设计了基于二级结构和残基-残基接触的选择策略来引导构象采样,分别用于提高算法在构象空间中探索近天然二级结构区域和合理结构的能力;此外,还设计了一个概率函数来平衡这两种选择策略;实验结果表明,该方案可以提高近天然态结构的采样能力[34]。在前期距离谱辅助片段组装研究的基础上,引入了残基间接触约束,提出了一种残基接触和距离谱耦合的结构预测方法CoDiFold[35]。设计了两个基于残基接触和距离谱的能量项,并将其融合到Rosetta 低分辨率能量函数score3 中;由两个残基接触联合的接触能量项用来约束构象,在基于接触的距离谱能量项中,利用接触信息来减弱或增强距离谱的约束;两个能量项的设计是为了缓解低分辨率能量函数的不准确性,提高模型能量与预测精度的相关性;针对搜索过程中容易陷入局部极小值的问题,设计了3 种不同的变异策略,以提高开发和勘探的性质。针对结构灵活的蛋白质结构loop 区域,提出了一种基于全局探索和loop 扰动的残基接触辅助的从头蛋白质结构预测方法CGLFold[10]。使用过滤后的残基间接触信息构建选择模型指导构象采样。在全局探索阶段,利用片段重组和片段组装大规模构象空间并生成近似天然态的拓扑结构;在loop 扰动阶段,设计了loop 区域特定的局部扰动模型,并通过差分进化算求解扰动量,进一步提高构象的精度。实验结果表明,loop 扰动可以对拓扑进行微小的调整,这种微小的调整不断累积最终产生可观的增益,进而显著提高预测模型的精度。由于能量力场的不完善,计算蛋白质折叠模拟中的数学最优解并不总是对应于最优结构,传统的构象采样算法难以跨越高能障碍物,容易陷入局部盆地。针对该问题,在最新的研究中课题组提出了两种多模态蛋白质结构预测方法[36‑37]。实验结果显示,通过多模态优化算法可以有效避免采样浪费或者采样不充足,并且能够采样到更多具有多样性的近天然态构象,从而显著提升结构预测的效率和精度。
随着残基接触和距离预测精度的不断提升,基于几何优化的蛋白质结构建模方法得到了广泛应用。这类方法没有采用片段组装等精巧的折叠方法,而是利用预测的contact 或distance 构建几何约束,通过CNS 或梯度下降能量极小化协议生成结构模型。CONFOLD[38]、RaptorX[39]和DMPfold[40]等方法将contact 或distance 以及其他约束送入CNS 中来生成模型;AlphaFold[41]和trRosetta[42]等方法将预测的distance 分布转化成蛋白质特定的统计势能函数,并与经典的能量函数相结合,通过梯度下降能量极小化协议生成模型。
3 蛋白质结构预测实验评测
3.1 相关的蛋白质数据库
PDB[43]蛋白质结构数据库由美国Brookhaven 国家实验室于1971年创建,由结构生物信息学研究合作组织维护,是最全的结构数据库,收录了通过实验方法测定的蛋白质结构。PDB 数据库中收集了蛋白质、多糖、核酸和病毒等生物大分子的三维结构数据,这些数据可通过X 射线单晶衍射、核磁共振和电子衍射等实验方法测定。通过互联网信息门户和可下载的数据档案可以访问大型生物分子(蛋白质、DNA 和RNA)的三维结构数据。
UniProt[44]数据库是收录信息最全面的蛋白质序列数据库,主要包括UniParc 序列归档库、Uni‑ProtKB 蛋白质知识库和UniRef 序列参考库。UniProtKB 知识库包含了蛋白质的序列数据和大量注释信息,分为由人工审阅和注释的Swiss‑Prot 数据库和计算分析的TrEMBL 数据库;UniRef 数据库按照序列相似度将UniProtKB 和UniParc 中的序列分为UniRef100、UniRef90 和UniRef50 三个数据集,可显著减小数据库大小,从而加快序列搜索的速度。
3.2 评价指标
均方根偏差(Root mean square deviation,RMSD)和TM‑score[45]是两种常用的计算目标结构与参考结构相似度的评价指标。RMSD 表示两个蛋白质结构经过结构的刚体旋转平移后计算原子间的平均距离,以Å 为单位,1 Å=10-10m,RMSD 值越小,表明两个结构越相似。通常主要考虑主链上Cα 原子间的RMSD。假设对于某个目标蛋白,考虑预测蛋白质模型P与实验测定结构P′的n个原子,RMSD计算公式为
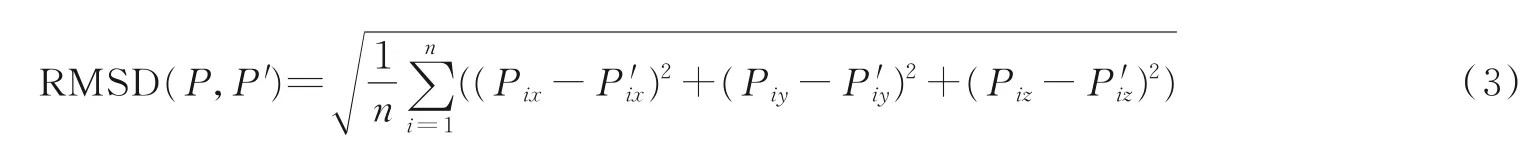
式中:(Pix,Piy,Piz)和分别表示模型P和结构P′第i个原子的三维坐标。
TM‑score 也是通过刚体旋转平移比对结构的相似度。不同于RMSD 的是,结构的局部差异对TM‑score 的影响较小。TM‑score 的大小不受蛋白质序列长度的影响,取值在(0,1]之间,其计算公式为[45]
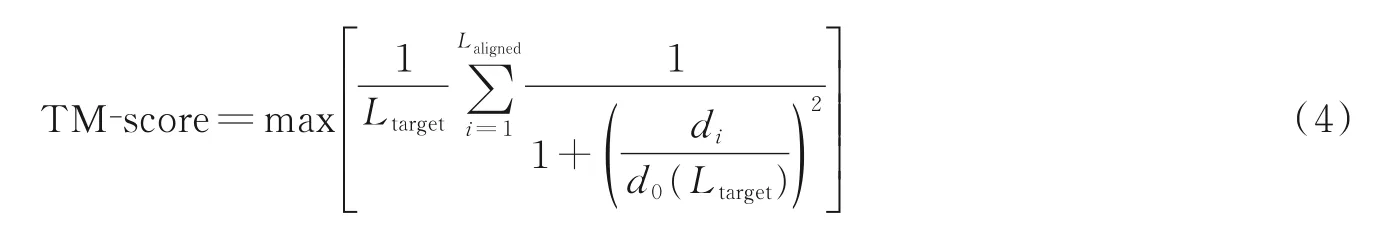
式中:Ltarget为目标蛋白的序列长度;Laligned为两个结构对齐区域的长度;d0为距离归一化参数,;di为第i个残基对间的距离。两个结构越相似,它们之间的TM‑score 越大;当TM‑score≥0.5 时,表明两个结构的拓扑形状大致相同[46]。
3.3 几种常见基于片段组装的蛋白质结构预测方法的性能分析与比较
为了真实反映近几年基于片段组装的蛋白质结构预测方法的性能,本节根据最新的基于片段组装的结构预测相关论文进行了方法描述,并对论文中的实验结果进行性能分析与比较。
CGLFold[10]是一种基于全局探索和loop 扰动的残基接触辅助的从头蛋白质结构预测方法。在全局探索阶段,利用片段重组和片段组装大规模构象空间并生成近似天然态的拓扑结构;在loop 扰动阶段,设计了loop 区域特定的局部扰动模型,并通过差分进化算求解扰动量,进一步提高构象的精度。如图2 所示,loop 扰动可以对拓扑进行微小的调整,这种微小的调整不断累积最终产生可观的增益,进而显著提高预测模型的精度。
MMpred[36]是一种distance 辅助的多模态优化采样方法。如图3 所示,在蛋白质结构预测过程中,能量模型的不准确性导致数学上的最优解不一定对应于天然态结构,而次优解或局部极小值解可能与之对应。MMpred 包括模态探测、模态维持和模态增强3 个阶段。在模态探测阶段,通过结构相似性快速评估模型来控制种群的多样性,在不同的低能量盆地中生成具有多样性的构象;在模态维持阶段,通过自适应聚类算法对种群进行划分,并调节蒙特卡罗模拟退火温度来实现模态的融合;在模态增强阶段,使用贪婪搜索策略加快模态收敛速度,并利用预测的残基间距离信息设计构象评分模型指导构象选择。
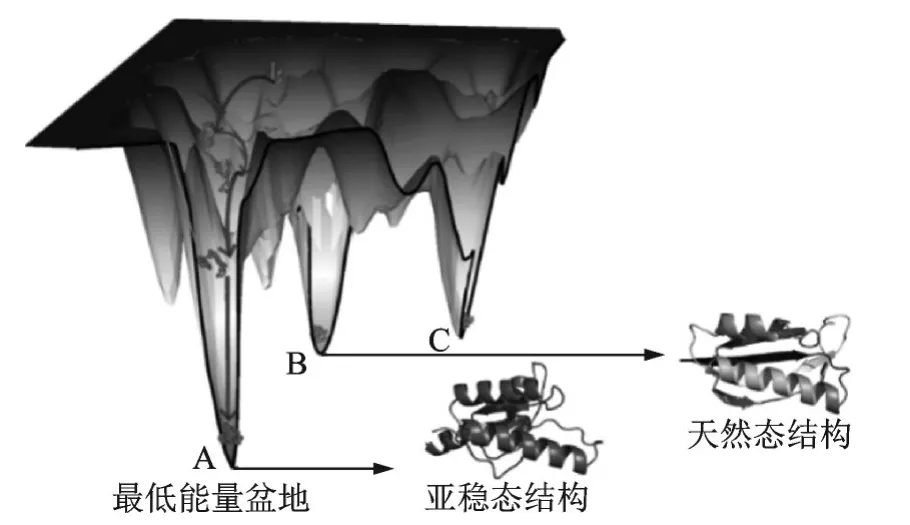
图3 蛋白质折叠的能量景观示意图Fig.3 Schematic diagram of protein-folding energy landscape
表1给出了CGLFold、QUARK、BAKKER ‑ ROSETTASERVER、MULTICOM_CLUSTER 和RaptorX‑Contact 在14 个CASP13 的FM 目标上的预测精度[10]。QUARK、BAKKER‑ROSETTASER‑VER、MULTICOM_CLUSTER 和RaptorX ‑ Contact 是CASP13 中4 种先进的服务器方法,其中QUARK 在FM 目标蛋白的搜索服务器组中排名第一。QUARK 和CGLFold 均是基于片段组装开发的算法,可以发现QUARK 和CGLFold 在这14 个FM 蛋白上取得了更高的平均预测精度。

表1 CGLFold、C⁃QURK、MULTICOM_CLUSTER、BAKER⁃ROSETTASERVER 和RaptorX⁃Contact在14 个CASP13 的FM 目标上的预测结果比较[10]Table 1 Prediction results comparison of CGLFold,C⁃QUARK,MULTICOM _CLUSTER,BAK⁃ER⁃ROSETTASERVER,and RaptorX⁃Con⁃tact on the 14 FM targets of CASP13[10]
表2 给出了MMpred 和Rosetta‑d(distance 约束的Rosetta 片段组装方法)在320 个非冗余基准测试蛋白上的平均预测结果[36]。MMpred 与Rosetta‑d 使用了相同的片段库、distance 约束和能量函数。可以发现在相同条件下,MMpred 取得了更高的预测精度。
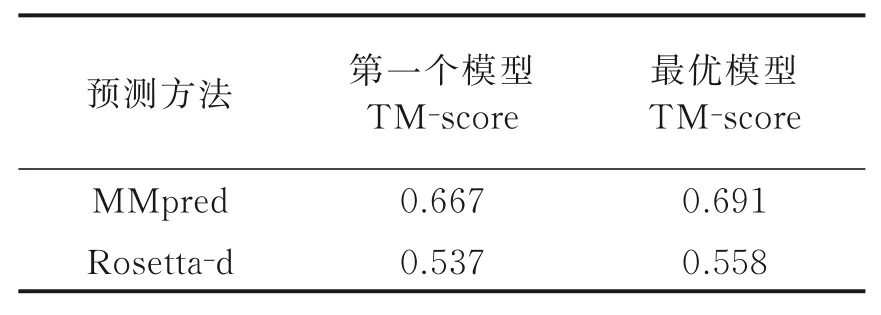
表2 MMpred 和Rosetta⁃d(距离约束的Rosetta)在320 个基准测试蛋白上的平均预测结果[36]Table 2 Average prediction results of MMpred and Rosetta⁃d(Rosetta with distance con⁃straints)on 320 benchmark proteins[36]
4 结束语
蛋白质三维结构的测定对疾病研究、诊断医疗和药物设计等有着重要的作用。然而,利用生物实验方法测定蛋白质结构耗时费力,代价极高。以计算机技术为手段实现蛋白质结构从头预测得到广泛关注。片段组装作为一种有效的插件式蛋白质构象空间优化技术,在蒙特卡洛构象优化算法中得到了广泛的应用。然而随着基于深度学习的残基间距离预测精度的不断提升,越来越多的方法直接采用几何优化方法来快速生成三维结构。为了进一步提升基于片段组装的蛋白质结构预测的性能,以下几个方面的研究方向是潜在的突破口。
(1)从已有研究成果来看,对于基于片段组装的蛋白质结构预测方法而言,构象空间采样仍然是一个瓶颈问题,尤其是随着蛋白质长度的增加构象空间呈几何倍数扩大。因此,设计高效的采样策略是提高算法效率和预测精度的关键之一。此外,片段组装将连续的二面角优化问题转换成了离散的实验局部结构的组合优化问题,虽然有效缩小了构象搜索,但也导致极有可能无法搜索最优解,并且随着蛋白质长度的增加这种影响会不断累计扩大。因此,如果能设计一个连续的二面角优化策略与离散的片段组装形成互补,将有望弥补片段组装这一固有缺陷。
(2)蛋白质能量模型不仅崎岖复杂,其构象搜索空间也十分庞大,这使得现有方法极易收敛到局部极值解。另外,即使搜索到全局最优解,能量模型的不准确性使得最优解不一定是最稳定的天然结构。进化计算社区的多模态优化方法,不仅能够发现全局最优解,而且可以获得更多样化的次优解,从而缓解能量模型的不准确性,提高搜索算法本身的稳定性(比如,全局最优解不一定对应于天然结构,某一个次优解可能更接近稳定的天然结构)。因此,基于群体的多模态优化方法是提高预测精度的重要保障。
(3)深度学习技术在蛋白质残基间距离预测中的成功应用使得蛋白质结构预测的精度取得了突破性进展,基于几何约束的能量极小化方法逐渐成为主流。然而,片段组装仍然具有其独特优势,既利用了已知蛋白质结构信息,又避免了同源建模方法高度依赖模板质量的缺陷,这使得片段组装方法能够正确预测具有未发现的整体拓扑结构的蛋白质结构。如果能够针对精细的残基间距离信息设计具有针对性的搜索算法,或是将能量极小化协议引入到构象采样过程中形成互补,可能会推动基于片段组装和基于几何约束能量极小化方法的进一步发展。
猜你喜欢
同位素(2022年6期)2022-12-30
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
肝博士(2022年3期)2022-06-30
物理学报(2022年10期)2022-06-04
海外星云(2021年9期)2021-10-14
食品与生物技术学报(2021年5期)2021-06-24
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
唐山师范学院学报(2020年6期)2020-04-16
唐山师范学院学报(2019年3期)2019-06-18
