
血液样本蛋白质组分析方法的比较研究
2021-09-14 04:37:16王智博王道平苗兰李瑛潘映红刘建勋
生物技术通报 2021年8期
王智博 王道平 苗兰 李瑛 潘映红 刘建勋
(1. 中国中医科学院西苑医院基础医学研究所,北京 100091;2. 中国农业科学院作物科学研究所,北京 100081;3. 北京市中药药理学重点实验室,北京 100091)
蛋白质组学技术作为基因组学和转录组学的补充,可用于基因组所表达蛋白的定性定量[1],在发现疾病潜在蛋白分子标志物和药物靶点以及探究蛋白作用机制等领域具有广阔的应用前景。蛋白质组学常以细胞、组织和血液为研究材料,其中血液样本因其微创易得且更能反应生物体的综合状态,目前广泛用于相关研究中[2-4]。
蛋白质组学研究的实验流程主要由样本制备、质谱分析、数据处理和生物信息学分析等环节构成。在这些研究环节中,属于样本制备范畴的预处理和酶切技术可显著影响蛋白鉴定率和数据重复性等后续质谱分析结果[5]。尽管当前应用广泛的超滤法样本制备技术简化了溶液内酶切的浓缩和除盐步骤,能减少样本损失,获得较高纯度肽段[6],血液样本的预处理和酶切技术仍需进一步完善。
血液样品的复杂构成特别是高丰度蛋白是影响蛋白质组学分析效果的主要因素[7]。研究表明基于抗体的亲和色谱吸附可有效去除血样中的高丰度蛋白[8]。基于不同的研究目的,血液样品可分成血清和血浆,其中血清在制备过程中已去除大部分纤维蛋白原,常用于蛋白质组学分析[9]。酶切技术在血液样本制备中的重要性也不容忽视。酶切前通常进行蛋白质变性处理,其中热变性较化学变性与质谱兼容性高,操作简便且不引入外来干扰物,可作为化学变性的替代方法[10]。Schniers等和Betancourt等[11-12]还报道在低浓度尿素溶液进行胰蛋白酶酶切时,可获得较好的重复性和深度的蛋白组覆盖。此外,多数胰蛋白酶切在37℃条件下进行,但Budnik等[13]报道在45℃条件下也可获得较好的酶切效率。有关酶切技术的优化有大量报道,这些改进技术值得制备血液样本时参考。
质谱技术是蛋白质组学的关键技术。Orbitrap质谱技术是现阶蛋白质组分析的主流技术之一,其获得碎片离子信息的方式常包括数据依赖性采集(data dependent acquisition,DDA)、非数据依赖性 采集(data independent acquisition,DIA)和平行反应监测(parallel reaction monitoring,PRM)3种模式[14]。DDA即经典的鸟枪法,是基于质谱的蛋白质组学实验的首选方法[15],该模式选择一级质谱上峰强度相对高的母离子进行碎裂,但母离子单同位素峰不一定是最高峰,低强度峰和共洗脱峰也较难鉴别。DIA解决了离子选择随机性的问题,不依赖预设参数,通过固定窗口或可变窗口进行高低离子交替采集,理论上重复性更高,相比DDA在质谱重复上潜力显著,但数据解析难度大[16]。Venable等[17]将DIA应用于酵母全细胞裂解物分析中,相比DDA-MS直接扫描定量获得了更高的信噪比、灵敏度、选择性和动态范围。近年来DIA谱图软件解析软件不断更新,极大提升了蛋白质组数据定量的准确度[18]。不同于DIA技术,PRM聚焦于目标肽段产物离子的监测[19]。PRM靶向蛋白质组学技术具有较高的精确度和重复性,但需要预先设定待检测前体离子,且检测数量有限[20],因此常应用于特定蛋白验证。Silva等[21]使用混合消化高密度脂蛋白(HDL)中的标记多肽进行DIA和PRM分析,发现两种模式均可获得较好的蛋白定量线性范围、准确度和精密度。
现有的血样制备分析方案较多,但发现生物标志物的进度仍旧缓慢[2]。血液样本包含重要的生物信息,其蛋白质组分析的意义重大。通过血样蛋白质组分析方法的比较研究,有望建立蛋白鉴定覆盖率较高和重复性较好的血液样本制备和分析方法,为进一步的蛋白质组学研究创造条件。
1 材料与方法
1.1 材料
1.1.1 动物 动物实验选用6周龄SPF级SD大鼠1只,体质量180 g,由北京维通利华实验动物技术有限公司提供,许可证号SCXK(京)2019-0001。取血前饲养于中国中医科学院西苑医院SPF级实验动物中心,温度(33±2)℃,相对湿度45%-65%。本研究通过中国中医科学院西苑医院动物伦理委员会批准(2018XLC003-2)。
1.1.2 试剂 水合氯醛购自国药集团化学试剂有限公司(批号30037517);色谱用水购自中国杭州娃哈哈有限公司;尿素(urea)购自美国昂飞公司(Affymetrix);胰蛋白酶(trypsin)购自普洛麦格 公 司(Promega);甲 酸(formic acid,FA)、二硫苏糖醇(dithiothreitol,DTT)、乙腈(ACN)购自美国赛默飞世尔公司(Thermo Fisher);碘乙酰胺(iodoacetamide,IAA)购自GE公司;碳酸氢铵(ammonium bicarbonate)购自Amresco公司。
1.1.3 仪器与耗材 Easy nLC 1000 纳升级液相色谱仪(美国Thermo Fisher公司);Q Exactive Plus 质谱仪(美国Thermo Fisher公司);预柱(富集柱,目录号HS-Trap-C-5U-5CM)和分析柱(直喷毛细色谱柱,目录号HS-Anal-C-5U-15CM)购自北京乐润峰科技有限公司(Beijing Happy Science Scientific Co.Ltd);CS5000型动物体重秤(上海奥豪斯仪器有限公司);ICE-CL31R型低温离心机(美国Thermo Fisher公司);-80℃超低温冰箱(美国Thermo Fisher公司);ProteoExtract Albumin/IgG Removal Kit(美国Merck公司,目录号122642)。
1.2 方法
研究流程见图1,采用基于超滤膜的过滤辅助法制备样品。使用维恩图(https://bioinfogp.cnb.csic.es/tools/venny/)进行定性结果重复性分析。主成分分析和数据可视化使用R软件包ggplot2_3.3.0和ggfortify_0.4.9。其他数据统计软件为Excel。
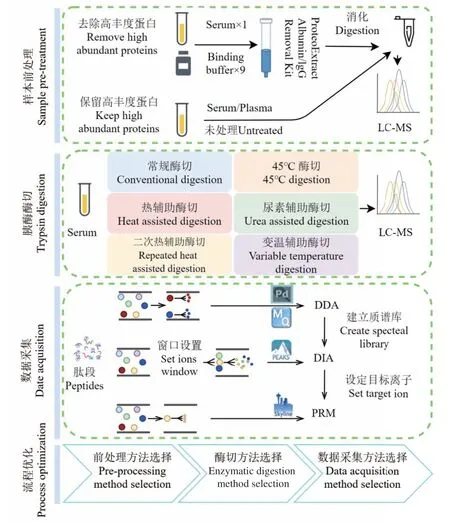
图1 实验流程Fig.1 Experimental flow
常规酶切方法、色谱质谱参数设置和数据库搜索方法参考文献方法进行[5]并稍作修改。流动相,A为纯水,B为CAN,均含0.1% FA(V/V)。流速设定420 nL/min,梯度洗脱设定:0.4 mL/min,3%-6% 3 min,6%-22% 78min,22%-100% 1 min,100% 8min。质谱分析在Q Exactive Plus质谱仪上完成。DDA采集模式的一级质谱分辨率70 000,扫描范围300-1 800 m/z,自动增益(automated guided cart,AGC)值3e6,注入时间(IT)50 ms。二级质谱的分辨率17 500,AGC值1e5,标准化碰撞能(normalized collision energy,NCE)27.0。毛 细管温度275℃,电喷雾离子源1900V,二级采集标准为强度前20的母离子进行Orbitrap检测。质谱原始数据分别采用Proteome Discoverer 2.1(Thermo Fisher) 和 MaxQuant 1.3.0.5(https://www.coxdocs.org/)对原始数据的RAW文件定性和定量,搜索引擎SEQUEST HT。用UniProt Rattus norvegicus 蛋白数据库(更新时间2020.11.1)进行检索。设定参数如下,高置信度,肽段长度设定6-144,最大漏切位点为2,前体离子质量容差±10 μmol/L,碎片离子质量容差±0.02 Da,固定修饰设定半胱氨酸碘乙酰胺化(carbamidomethy/+57.021 Da),可变修饰选择甲硫氨酸氧化(oxidation/+ 15.995 Da)和N-乙酰化(Acetyl/+42.011 Da),蛋白和多肽定性假阳性率FDR为1%。每个样本的3次质谱重复合并检索,得到蛋白定性数据以Excel格式导出。
1.2.1 血样预处理 血清采集:采集全血于试管中,4℃静置30 min待凝固后离心(1 500×g,10 min),取上清置于EP管中,-80℃保存。血浆采集:采集全血于医用血常规管(EDTA抗凝)中,4℃离心(1 500×g,10 min),取最上层置于EP管中,-80℃保存。血清去除高丰度蛋白处理:-80℃保存的血清解冻后,参考ProteoExtract Albumin/ IgG Removal Kit说明书去除高丰度蛋白。
3种预处理的大鼠血液样本按照常规方法进行蛋白组样品制备和质谱检测,步骤如下:样本的初始体积均为50 μL,浓缩后置于超滤管采用8 mol/L尿素洗涤3次,终浓度10 mmol/L DTT还原(37℃,30 min),8 mol/L尿素洗涤3次,终浓度20 mmol/L IAA烷基化(37℃,30 min),8 mol/L尿素洗涤3次,采用50 mmol/L NH4HCO3置换尿素后加入2 μg Trypsin进行酶切(37℃,16 h),离心(14 000 r/min,10 min)取下层液用于质谱分析。利用数据处理软件比较血浆、血清和去除高丰度蛋白血清的蛋白鉴定数、肽段数、谱图匹配数和蛋白定量重复性。
1.2.2 胰蛋白酶切(protein digestion) 取50 μL样本用于酶切处理,还原烷基化步骤同上,8 mol/L尿素和50 mmol/L NH4HCO3洗涤,最终得到约20 μL样品用于酶切。
1.2.2.1 常规酶切(conventional digestion,CD) 采用常规方法进行还原、烷基化和洗涤,加入2 μg Trypsin,37℃恒温水浴16 h,加入0.1%(V/V)甲酸水溶液终止酶切,更换接收管,离心(12 000 r/min,20 min),离心液用于质谱分析。
1.2.2.2 45℃孵育(45℃ digestion,45D) 按常规酶切方法处理样品,孵育温度设为45℃。
1.2.2.3 热辅助酶切(heat assisted digestion,HD)样品还原前加入50 mmol/L NH4HCO3定容到1 mL,在密闭的微型离心管中加热(95℃,10 min),转移到冰水浴中降温终止变性过程,浓缩,余下步骤同CD法。
1.2.2.4 二次热辅助酶切(repeated heat assisted digestion,RHD) 样本还原前加入50 mmol/L NH4HCO3定容到1 mL,在密闭的微型离心管中加热(95℃,10 min),降至室温后浓缩,加入1 μg酶,37℃孵育3 h,二次加热样品(95℃,10 min),降温后再加入1 μg酶,37℃孵育13 h。
1.2.2.5 尿素辅助酶切(urea assisted digestion,UD) 采用常规方法进行还原、烷基化和洗涤,酶切前加入终浓度为1 mol/L的尿素,余下步骤同CD法。
1.2.2.6 变温酶切(variable temperature digestion,VD) 采用常规方法进行还原、烷基化和洗涤,酶切前将样品管置于80℃水浴锅,待水温降至60℃后加入含2 μg Trypsin的酶液,随后转入37℃水浴锅孵育16 h。
1.2.3 质谱数据采集
1.2.3.1 数据依赖性采集(DDA) DDA条件设定及数据处理主要参考文献方法设定[5],其余参数设置见前述。
1.2.3.2 非数据依赖性采集(DIA) 肽段液相分析方法同上,质谱设定为DIA采集模式,一级质谱设定为Full MS,二级质谱扫描设定60个固定扫描窗口,分辨率、扫描范围、AGC值、注入时间等参数同DDA。将DIA数据导入到PEAKS studio 8.5进行定性和定量,数据采集模式设定为DIA,首先使用DDA采集模式的raw文件构建DIA检索的蛋白谱图库,再对数据进行从头序列(de novo sequencing) 分析,肽段长度、离子质量偏差、假阳性率、固定修饰和可变修饰等参数同DDA,每个样本的3次质谱重复合并检索,得到蛋白定性数据以Excel格式导出。
1.2.3.3 平行反应监测(PRM) 肽段液相分析方法同DDA,质谱设定为PRM采集模式,在设置菜单中选择离子对设置,将母离子和子离子质量分析仪设置为Orbitrap,分析时间90 min,正离子检测,MS1 分辨率为70 000(200 m/z),MS/MS 分辨率为17 500(200 m/z),AGC、NCE、IT等参数同DDA。将PRM定量的21个靶蛋白导入Skyline软件(https://skyline.ms/)中,选择肽段设置,添加背景蛋白组数据库文件,根据谱库中的离子信号选择蛋白质定量的多肽,从Skyline导出包含保留时间的相关肽列表,并手动检查靶蛋白的每个肽的定量结果。
1.2.4 血清蛋白组分析流程优化 根据上述血样蛋白质组分析方法的比较研究结果,采用优化的前处理、酶切和数据采集方法构建适宜的血样蛋白组分析流程。
2 结果
2.1 血样前处理方法比较
每个样品进行3次重复实验,其中血浆样本平均检测到321组蛋白,重复鉴定的蛋白占比66.2%;保留高丰度蛋白血清平均检测到346组蛋白,重复鉴定的蛋白占比64.4%,相较于血浆增加了16组蛋白鉴定数;去除高丰度蛋白血清平均检测到377组蛋白,重复鉴定的蛋白占比69.4%,相较于保留高丰度蛋白血清增加31组蛋白鉴定数(图2-A)。进一步的蛋白定性分析结果显示,去除和保留高丰度蛋白血清样品的蛋白鉴定数和二级谱图数无显著性差异,但去除高丰度蛋白样品的肽段和谱图匹配数(peptide spectrum matches,PSMs)均高于保留高丰度蛋白样品(图2-B)。3种血液样本定量数据的主成分分析显示,3种血样的组间分离度明显较高,但每种血样的3个重复样本彼此相近,如图2-C所示。
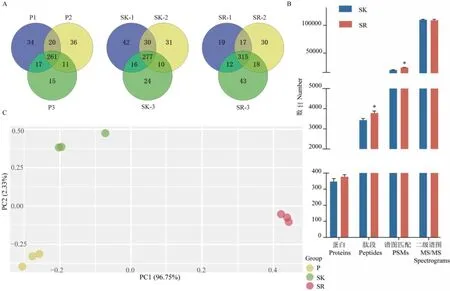
图2 三种预处理方法制备血样的质谱结果比较Fig. 2 Comparison of mass spectrometry results of blood samples prepared by 3 pre-processing methods
2.2 酶切方法比较
对不同酶切样本质谱鉴定的蛋白数、肽段数、谱图匹配数进行统计,如图3-A所示,45D组所得蛋白鉴定覆盖率低于CD组;HD组鉴定到的蛋白数、肽段数和谱图匹配数较其他组高,其中肽段数和谱图匹配数相比CD组有显著性差异,低标准差表明HD组重复性更高;RHD组蛋白鉴定数较CD组低,但识别的肽段数高于CD组;UD组显示蛋白数、肽段数和谱图匹配数相对CD组低。而VD法所得蛋白和肽段鉴定率较CD组高,无显著性差异;另外,VD组各项指标显示出较高的标准差。分析每组样品的3次重复制样,45D组重复率为69.7%,高于其他5组图;HD组3次重复鉴定到的共有蛋白数最多,为288个(图3-C)。比较各组样品的遗漏酶切位点数,VD组酶切位点遗漏率为51.22%,较其他组最低,而RHD组和UD组具有相对较高的漏切率(图3-B)。另外,分别统计了蛋白序列覆盖度在20%-35%、35%-50%和>50%的区间分布(图3-D),发现在20%-35%和>50%的区间,VD组和HD组相比其他4组具有更高的蛋白序列覆盖度,其中HD组具有更低的标准差,表明HD组蛋白被酶解的更充分;而在区间35%-50%中,UD组和RHD组有最高的肽段分布。对定量结果的主成分分析显示,组间交叉明显,其中45D组在主成分1(PC1)的分析中差异最小,RHD组次之;HD组在主成分2(PC2)的分析中差异最小,VD组次之(图3-E)。
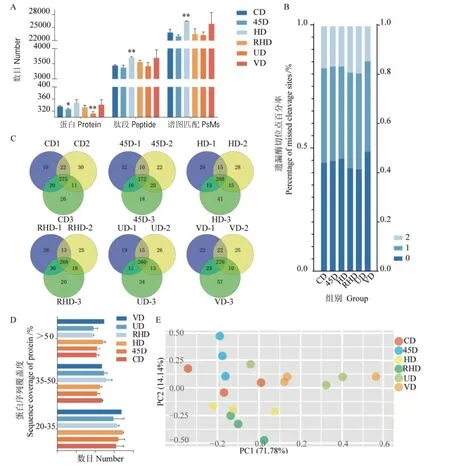
图3 血清蛋白不同酶切方法质谱结果比较Fig. 3 Comparison of mass spectrometry results of serum proteins with different enzymatic digestion methods
2.3 DDA和DIA数据采集模式结果分析
血清样品再DDA模式下单次质谱分别鉴定到338、346、359个蛋白;DIA模式下以DDA的raw文件为背景库单次质谱分别鉴定到371、368、369个蛋白,单次质谱DIA模式更佳(图4-A)。将3次质谱重复蛋白定性结果合并后剔除重复值,发现DDA和DIA模式分别有422和420个蛋白鉴定数,但DIA具有更小的标准差,且鉴定到的共有蛋白数占总蛋白的比例分别为68.2%和75.9%(图4-C)。因为质谱分析中普遍存在蛋白数据缺失的状况,缺失率可作为反映数据采集稳定性和重复性的指标之一,因此根据样本中蛋白的检出度,分别统计了DDA和DIA的3次质谱重复蛋白定性数据的缺失值。在DDA模式下,定性数据缺失率分别为26.41%、26.66%、25.12%;在DIA模式下,缺失率分别为11.45%、12.17%、11.93%图(4-B)。进一步筛选3次质谱重复鉴定到的肽段信息,发现DDA和DIA分别鉴定到共有肽段数为3450和1569,肽段CV值的均数分别为34.65%和22.12%,CV值低于20%的肽段数分别占比29.68%和67.81%图(4-D)。
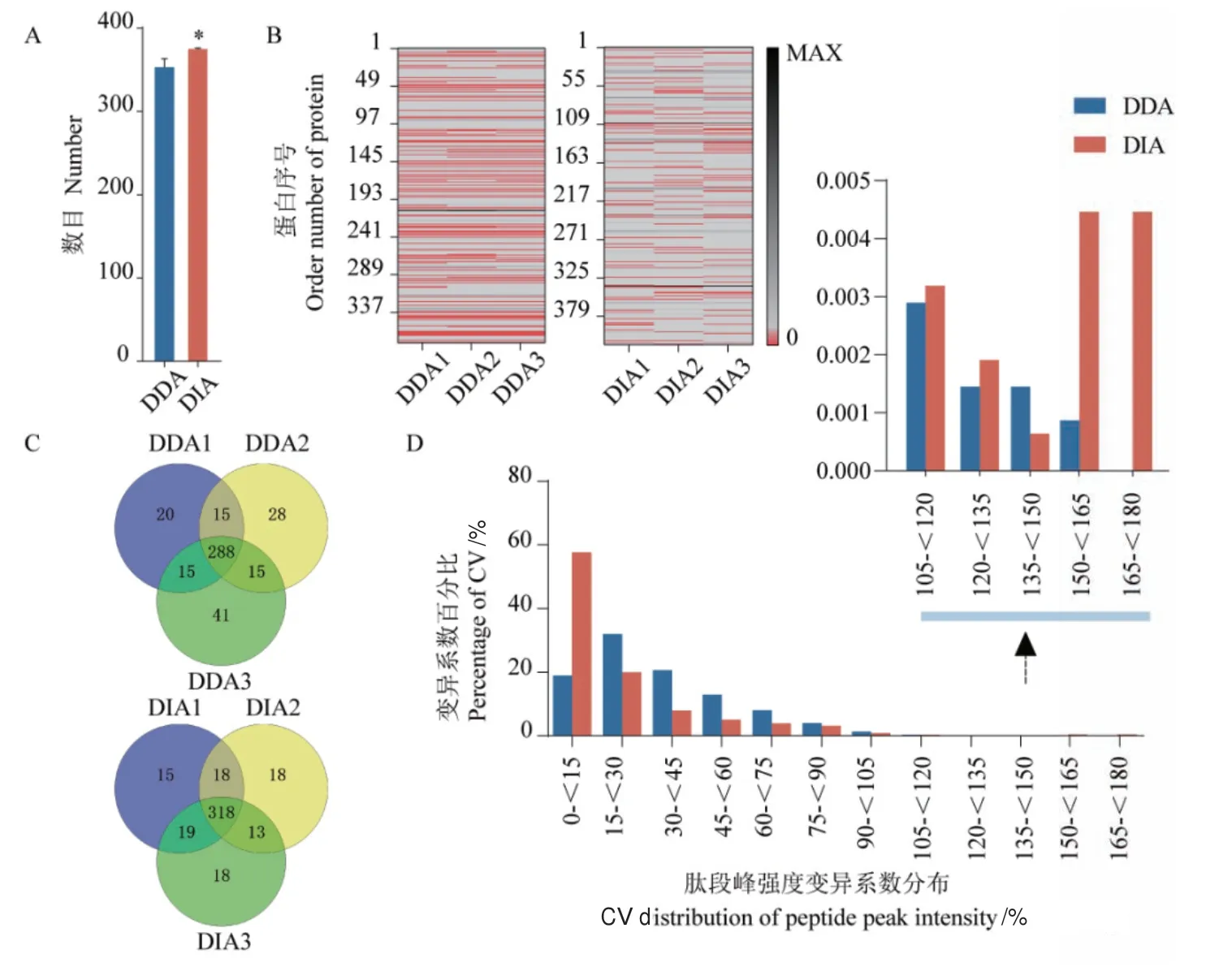
图4 DDA和DIA数据指标比较Fig. 4 Comparison of DDA and DIA data indices
2.4 DIA和PRM数据采集模式结果比较
选取不同丰度梯度的蛋白比较DIA和PRM在蛋白组中的定量分析能力,通过各特异肽段峰面积计算3次质谱重复的CV值。如表1所示,两种检测方法中CV值低于20%的比例分别为42.85%和47.16%,通常筛选特异肽段信息完成蛋白定量,因此多数情况下PRM相对DIA定量更有优势,在一些蛋白的定量中DIA也展示出良好的定量重复性。
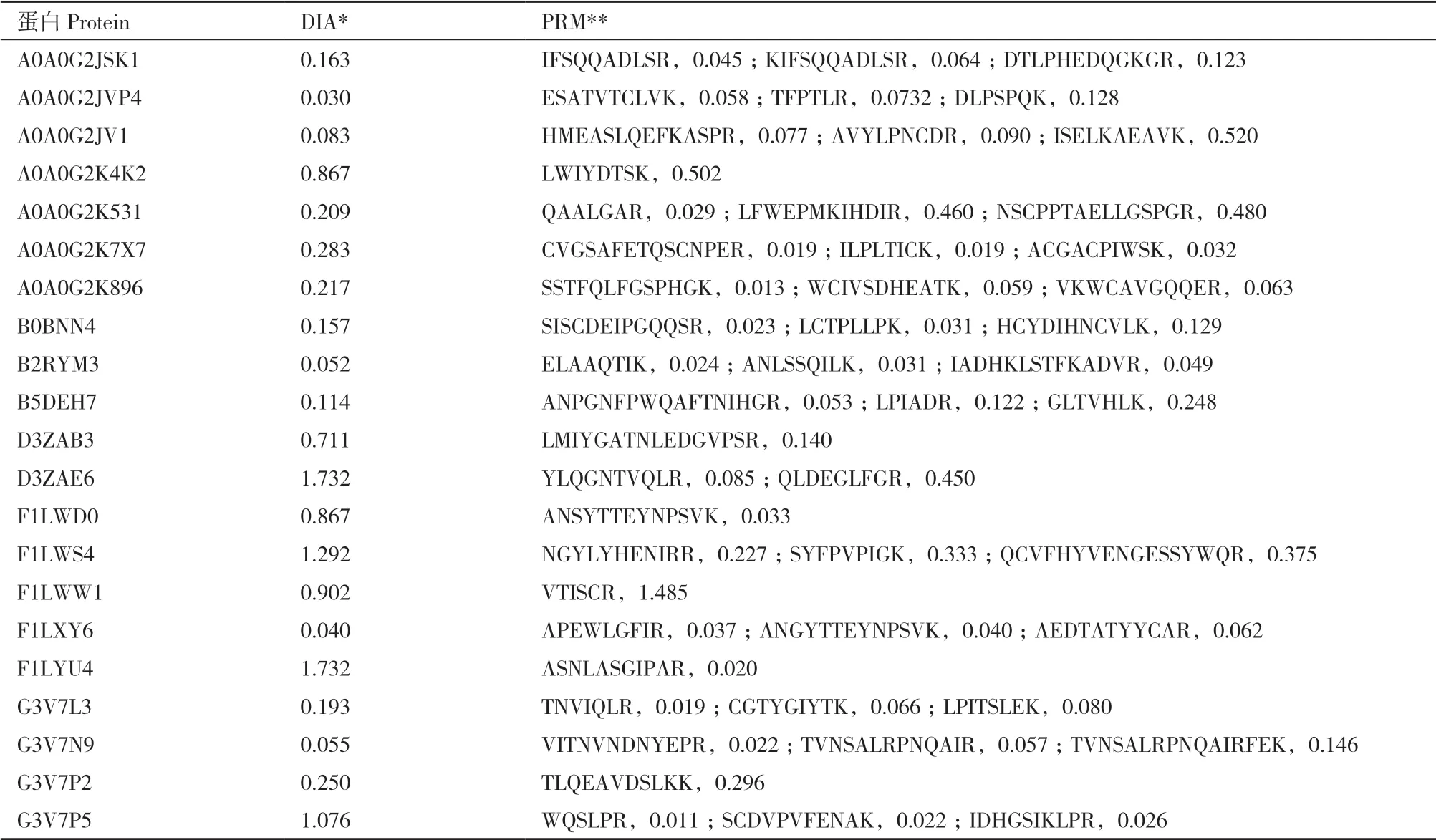
表1 DIA蛋白和对应肽段PRM 3次重复定量CV值分析Table 1 Quantitative analysis on the coefficient of variation of protein from DIA and corresponding peptides from PRM in 3 repeats
2.5 血样蛋白质组学流程优化
血清去除高丰度蛋白,经热辅助结合变温酶切法酶切,最后采用DDA数据采集模式分析,3次重复试验分别定性到490、490、504个蛋白,鉴定总蛋白数590个,共有蛋白占比69.8%(图5-A);定量CV值低于20%的蛋白占比71.10%(图5-B)。
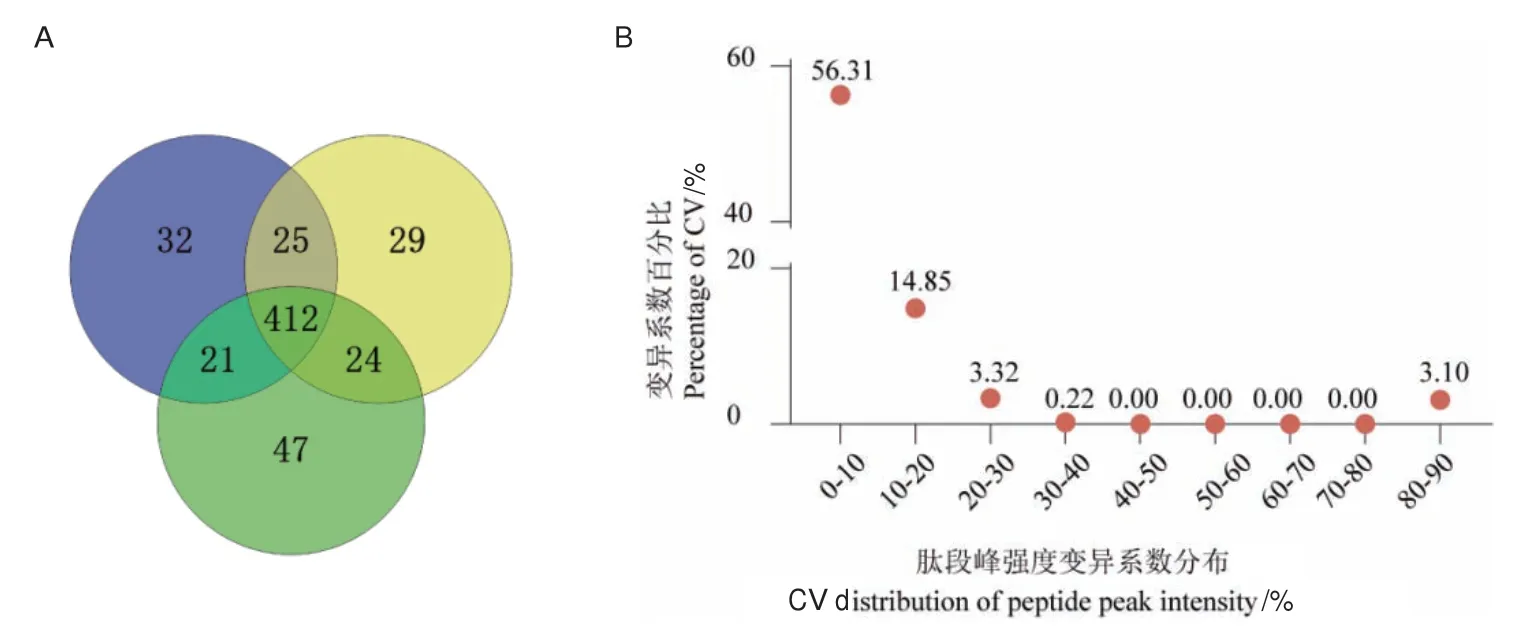
图5 基于优化流程分析的血样蛋白组定性和定量结果Fig.5 Qualitative and quantitative results of blood sample proteome based on optimized workflow
3 讨论
3.1 样本前处理结果比较
血样蛋白质组学研究首要环节是样本的前处理。定性结果显示血浆蛋白鉴定数最少,保留高丰度蛋白的血清重复性最低,去除高丰度蛋白的血清蛋白鉴定数和重复性最高,提示血清去除高丰度蛋白可提高蛋白鉴定覆盖率和可靠性。在相同的色谱质谱条件和数据分析模式下,尽管两种血清样品的蛋白鉴定数和二级谱图数差异不显著,但去除高丰度蛋白后肽段数和谱图匹配显著数增加,证实血清去除高丰度蛋白更适合蛋白质组学研究。定量结果主成分分析显示血浆和去除高丰度蛋白的血清组内重复性较好,其中去除高丰度蛋白的血清3次重复具有最高的聚合度,表明去除高丰度蛋白处理在血清样本制备中具有一定的合理性,血清去除高丰度蛋白可获得更高的蛋白鉴定数和重复率。综合比较大鼠血样前处理样品的质谱鉴定结果,基于研究效率考虑,后续酶切和数据采集方法比较可采用保留高丰度蛋白血清为样本,但构建优化的血液样本蛋白质组分析流程时宜采用去除高丰度蛋白血清。
3.2 酶切方法比较
酶切是样本制备的重要环节,某些蛋白由于存在二硫键和特定空间构象,需要通过化学物质或者加热处理暴露酶切位点,酶切温度、缓冲液构成等条件同样影响酶切效率。本研究比较了常规酶切(CD)、45℃孵育(45D)、热辅助酶切(HD)、二次热辅助酶切(RHD)、尿素辅助酶切(UD)和变温酶切(VD)六种方法,发现酶切效率存在差异。HD法鉴定到的蛋白数、肽段数和谱图匹配数较其他组高,与前人热变性后肽段鉴定数增加的结果保持一致[10],表明胰蛋白酶酶切前高温变性更适用于血清样本蛋白质组分析。胰蛋白酶活性受温度影响较大,多数文献在37℃下酶切,但也有45℃的报道[13]。45D法相对CD法在蛋白数、肽段数和谱图匹配数上较低,说明胰蛋白酶在37℃孵育比45℃酶切效果更佳。RHD相比CD法蛋白数低但肽段数高,原因可能是酶切不完全,从而降低蛋白鉴定率。此外,UD法相对CD法蛋白检出率低。通常蛋白酶切存在一到多个位点的遗漏酶切,过高的酶切位点遗漏率会降低蛋白定量的精密度和准确度[22]。VD法酶切位点遗漏率最低,表明酶切更充分,但标准差较高,推测原因为在可耐受的范围内瞬间加入酶液,在温度变化的窗口区间酶活性激发所致,但实际操作中温度难以精确控制,从而导致VD法3次重复实验稳定性差。此外,HD和VD法在在20%-35%和>50%的区间内也显示出更高的蛋白序列覆盖度,表明两种方法得到的血清蛋白质组定性结果更可信和准确。6种酶切方法蛋白定性重复性差异不明显,其中45D法定性重复率略高,但HD法可获得更高的共有蛋白数,虽然HD法和VD法的定性重复性不突出,考虑到蛋白鉴定数、漏切位点和蛋白序列覆盖度,这两种方法更适用于血清蛋白质
组分析。除了上述基于定性数据的分析外,定量结果的主成分分析显示45D和RHD组在PC1(71.78%)的分析中差异较小,而HD和VD组在PC2(14.14%)的分析中差异较小,表明CD法和UD法的定量重复性相对较低,45D和RHD更适合血清蛋白质组定量分析。综合6种酶切方法定性定量结果分析,热辅助酶切配合变温酶切更适用血液样本蛋白质组学研究。
3.3 DDA与DIA比较
3次质谱重复蛋白鉴定数的平均值显示DIA具有更高的蛋白鉴定数,表明DIA在单次进样中更有优势。在蛋白定性数据中,DIA也展示出更好的重复性,合并后剔除重复值显示DDA和DIA蛋白鉴定数差异不明显,表明在多次进样的情况下,两种方法的蛋白定性能力相差不明显。缺失值统计结果显示DIA比DDA平均每组数据完整度高出14.21%,提示DIA具有更高的稳定性和重复性,多数实验已经证明相同结果[23]。另外,若设定0-15为可信区间,DIA在蛋白定量上显示出更低的变异系数。因此DIA更适用于筛选血样中的差异表达蛋白,同时,标记结合靶向定量因其更高的准确度、灵敏度和重现性已广泛应用于蛋白翻译后修饰、蛋白互作等领域[24-25]。研究中DIA仍需参考DDA建立的数据库进行检索,因此DDA在通量和便捷度上优势明显。综上可得,DIA重复性好且单次质谱鉴定蛋白数高但DDA操作更简便。
3.4 DIA与PRM比较
DIA和PRM是基于Orbitrap质谱的常用靶向定量方法,选取的21组蛋白或相应肽段CV值显示,多数特异肽段在PRM定量的CV值低于DIA中对应蛋白定量的CV值,提示PRM具有更高的稳定性,但在实际研究中监测离子数目有限,更适用于候选生物标志物的验证,DIA和PRM均可用于靶向蛋白质组学分析但PRM结果稳定性相对较高。
3.5 流程优化
去除血样高丰度蛋白有多种方法,评估各种试剂盒去除高丰度蛋白效率的研究也较多[26-27],本研究采用ProteoExtract Albumin/IgG Removal Kit去除血清高丰度蛋白,可获得更高的蛋白鉴定数和重复率。常规酶切方法使用广泛,但也有很多文献报道优化的酶切方法,如Lys-C/Trypsin顺序酶切[28]、微波和红外辅助酶切[29]等。本研究发现热辅助酶切配合变温辅助酶切可获得更好的质谱结果,更适用于血样蛋白组研究。在数据采集模式上,DDA和DIA各有优势,DIA因其较高的重复性和稳定性,单次质谱鉴定蛋白数高,而DDA无需预先建立谱图库,可直接通过蛋白序列库搜索从而完成蛋白的定性定量。PRM定量在多数情况下具有较高的稳定性,但单次进行靶向分析的蛋白数目有限,可用于对目标蛋白的相对和绝对定量。
综上所述,进行血样蛋白质组分析时,宜去除高丰度蛋白,采用热辅助酶切结合变温酶切法和DDA数据采集模式进行质谱分析,定量结果还可用PRM技术进一步验证(图6)。
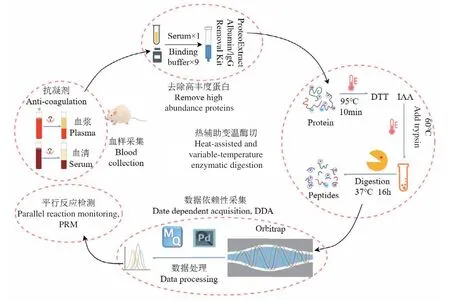
图6 血样蛋白质组样品制备和分析流程Fig. 6 Sample preparation and analysis workflow for blood proteome
4 结论
比较血样蛋白质组分析方法发现,血清样本去除高丰度蛋白后,经热辅助结合变温酶切法酶切,采用DDA数据采集模式进行质谱分析,可获得较高的蛋白鉴定覆盖率和重复性较好的定性定量结果,利用PRM技术还可对定量结果进行后续验证。基于血液样本蛋白质组学分析方法的比较研究,建立了优化的制备和分析流程,为进一步的蛋白质组学研究创造条件。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30 06:39:48
昆钢科技(2021年2期)2021-07-22 07:46:56
东坡赤壁诗词(2021年1期)2021-03-24 18:25:36
职工法律天地(2018年12期)2018-01-22 22:55:10
水利信息化(2017年4期)2017-09-15 12:01:21
护士进修杂志(2017年1期)2017-02-28 19:49:40
当代化工研究(2016年5期)2016-03-20 16:21:35
甘肃畜牧兽医(2016年20期)2016-03-12 03:59:00
四川畜牧兽医(2014年9期)2014-12-31 12:30:58
特产研究(2014年4期)2014-04-10 12:54:22
