
可抑制1,2-丁二醇生成的高性能草酸二甲酯加氢B-Cu/MS催化剂的研究
2021-09-14 06:48:22俞金山刘甜甜艾培培
天然气化工—C1化学与化工 2021年4期
俞金山,冯 翀,刘甜甜,张 莉,艾培培,黄 伟
(1.太原理工大学 化学化工学院,山西 太原030024;2.阳泉煤业(集团)有限责任公司化工研究院,山西太原030021;3.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原030024)
乙二醇(EG)作为一种广泛应用于聚酯、润滑剂、增塑剂、非离子表面活性剂和防冻剂等众多化工行业的原材料,在国内外具有较大的市场需求[1,2]。目前主要采用煤作为制备EG的原料,煤先经气化后生成合成气,然后经三种工艺路线(直接法、烯烃法和草酸酯法)制得EG。其中草酸酯法制备EG作为一种绿色、原子经济性高的生产技术,实现了煤炭资源的高效、清洁利用,引起了国内研究人员的广泛关注[3-8]。草酸酯法制EG主要工艺过程包括CO氧化偶联制草酸二甲酯(DMO)工艺和DMO加氢制EG工艺。其中,DMO的制备已然成熟并成功工业化。DMO加氢制EG过程中仍存在一些问题:(1)在反应温度下,Hüttig温度较低的活性铜物种易烧结失活,导致催化剂稳定性差[5-11];(2)高温下易发生Guerbet反应,生成难以从产物EG中分离的副产物1,2-丁二醇(1,2-BDO),影响EG的产品质量[5]。因此,如何在结构设计和合成探索的基础上开发高效铜基催化剂,从而实现DMO高选择性加氢制EG并降低或避免副产物1,2-BDO的生成对该工艺至关重要[12,13]。
铜基催化剂因其C=O键氢化活性较高和C-C键解离吸附能力较低被广泛应用于DMO加氢反应中[14,15]。研究人员对SiO2、Al2O3、ZnO和La2O3等不同载体负载的无铬铜基催化剂进行了广泛研究[16,17]。其中,SiO2载体因其中性特性及与Cu间的强相互作用,使得Cu/SiO2催化剂表现出稳定性较高的催化加氢性能[5]。研究发现,难分离副产物1,2-BDO是DMO加氢反应中EG和乙醇(EtOH)的反应产物,该反应为Guerbet反应[4,18-20]。Guerbet反应是EtOH、丙醇和丁醇等醇缩合而发生的二聚反应,其反应机理主要基于三个连续的反应路径[19]:(1)醇脱氢生成相应的醛;(2)生成的醛在碱性位的催化下进行羟醛缩合;(3)不饱和缩合产物加氢生成二聚醇。该反应机理说明通过调变催化剂的酸碱性抑制副产物1,2-BDO的生成在理论上是可行的。目前,部分研究人员在通过调控催化剂酸碱性抑制C3-C4OH(副产物丙醇、丁醇和丙二醇、丁二醇的统称)方面做了一些初步工作。Song等[20]通过在催化剂中添加铝组分减少铜基催化剂的碱性位,抑制C3-C4OH生成的同时提高了EtOH选择性,且当EG代替DMO作为原料时,C3-C4OH的形成受到显著抑制。Wang等[4]利用硅烷偶联剂对Cu/SiO2表面分离出的羟基进行了后接枝,羟基被覆盖后,碱性位点密度和强度均显著降低,从而降低了C3-C4OH的选择性,提高了EG的选择性。因此,副产物1,2-BDO完全可以通过调控催化剂酸碱性的方式从源头上进行抑制。
综合考虑,本文采用尿素协助沉淀法制备Cu/MS催化剂前驱体,并通过超声辅助浸渍法引入硼物种,以期通过弱酸性氧化硼的引入同时实现DMO催化加氢稳定性的提高和难分离副产物1,2-BDO的抑制。
1 实验部分
1.1 实验材料与试剂
正硅酸乙酯(TEOS,AR)、十六烷基三甲基溴化铵(CTAB,AR)、氨水(25%~28%,AR)、尿素(AR)、硼酸(AR),购自天津市科密欧化学试剂有限公司;无水乙醇(AR),购自天津市大茂化学试剂厂;Cu(NO3)2·3H2O(AR),购自上海麦克林生化有限公司。
1.2 催化剂制备
1.2.1 MS载体的制备
采用水热合成法制备MS载体:将TEOS、CTAB、水和氨水按1:0.2:160:0.4(物质的量比)混合溶解,并剧烈搅拌至产生白色凝胶;将白色凝胶装入水热反应釜100℃下加热48 h,冷却至室温后离心洗涤至中性,100℃干燥后经550℃下焙烧6 h,即得到MS载体。
1.2.2 Cu/MS前驱体的制备
采用尿素协助沉淀法制备Cu/MS前驱体:将3.40 g Cu(NO3)2·3H2O、5.85 g氨水(25%~28%)、1.50 g尿素及2.10 g MS载体依次加入到装有去离子水的圆底烧瓶中,混合溶解后80℃剧烈搅拌4 h,去离子水洗涤沉淀至中性,100℃干燥,即得到天蓝色的Cu/MS前驱体。
1.2.3 Cu/MS和xB-Cu/MS催化剂的制备
采用超声辅助浸渍法制备xB-Cu/MS催化剂,其制备步骤如下:首先,在超声辅助下将一定量的硼酸水溶液滴加到Cu/MS前驱体中,真空老化2 h,100℃真空干燥过夜,然后经400℃焙烧4 h即得铜理论质量负载量为30%的系列催化剂。该系列催化剂命名为xB-Cu/MS(x=1,2,3,4),其中x代表硼的理论含量。将Cu/MS前驱体在400℃下焙烧4 h,得到Cu/MS催化剂。
1.3 催化剂表征
采用美国康塔仪器QDS-30物理吸附仪表征催化剂的比表面积及孔结构。称取一定量催化剂放于样品管中,升温至200℃并脱气4 h,然后在液氮温度(77 K)下进行吸/脱附实验。采用BET和BJH方法测得催化剂相关织构参数(比表面积、孔径、孔容等)。
采用日本理学D/max-2500型X射线衍射仪表征催化剂XRD衍射特征,Cu Kα射线源(λ=0.15406 nm),扫描范围为5°~90°,速率为8(°)/min。
TEM表征采用日本JEOL公司的2010型高分辨透射电子显微镜测试,工作电压200 kV。室温下将催化剂样品在EtOH中超声分散30 min,得到的溶液滴在微栅上进行透射电镜观察。
H2-TPR表征采用天津先权TP-5000型多用吸附仪测试。在装有催化剂的测试管中通入惰性气体升温至150℃,预处理60 min;降温至50℃,惰性气体吹扫30 min后切换为H2/Ar气体进行分析,升温速率为10℃/min。
NH3-TPD表征采用天津先权TP-5000型多用吸附仪测试。在装有催化剂的测试管中通入惰性气体升温至150℃,预处理60 min;降温至50℃,通入待吸附气体(NH3,30 mL/min)并维持30 min后,用惰性气体吹扫30 min后进行分析,升温速率为10℃/min。
XPS表征采用Thermo ESCALAB 250XL型能谱仪测试,Al kα辐射(hv=1486.6 eV),结合能的计算以C 1s(284.6 eV)为基准,测量误差为(±0.05)eV。
1.4 催化剂性能测试
DMO加氢反应在内径为8 mm的石英管固定床反应器中进行,将0.5 g催化剂(40~60目)置于石英管缩口上端恒温区。催化剂首先在300℃、H2气氛下预还原3 h,随后将反应器温度调至反应温度,反应器压力升至2.5 MPa,原料液由高压输液泵输入反应器。液相产物经低温循环冷却泵冷凝后,由岛津GC-2014气相色谱进行离线分析,采用FID检测器,DB-624毛细管柱。
采用内标法对反应物及产物进行定量分析,催化剂的转化率及选择性由式(1)、式(2)进行计算:


式中,XDMO为DMO转化率,%;nin,DMO为原料液中DMO物质的量,mol;nout,DMO为产物中DMO物质的量,mol;Si为产物i的选择性;ni为产物i的物质的量,mol;ni,DMO为转化的DMO的总物质的量,mol。
2 结果与讨论
2.1 Cu/MS和xB-Cu/MS催化剂在DMO加氢反应中的活性和稳定性
在180℃、2.5 MPa、质量空速(WHSV)0.4 h-1和n(H2)/n(DMO)为100的反应条件下,研究了Cu/MS催化剂以及xB-Cu/MS催化剂的催化性能,结果如表1所示。由表1可知,Cu/MS催化剂表现出较佳的催化加氢性能,DMO转化率达100.00%、EG和EtOH选择性分别为94.13%和4.93%,但仍有部分难分离副产物1,2-BDO产生(选择性为0.63%)。
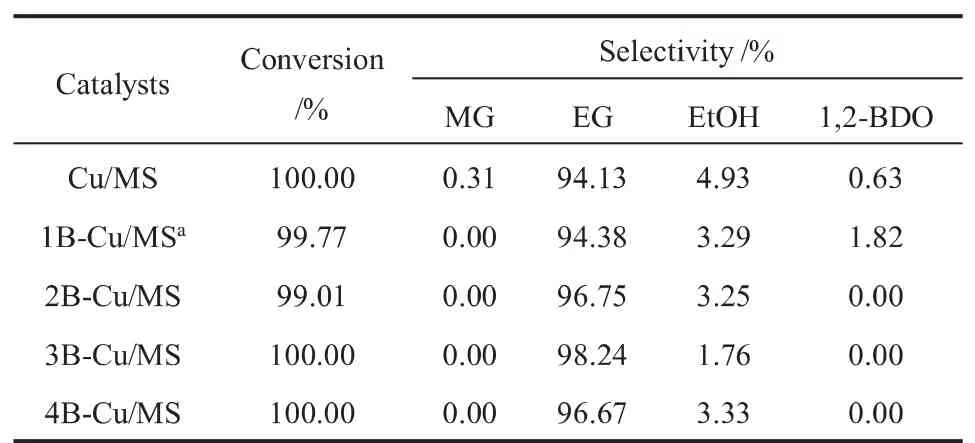
表1 Cu/MS和xB-Cu/MS催化剂的催化性能Table 1 Catalytic performance of Cu/MS and xB-Cu/MS catalysts
实际生产中DMO加氢催化剂的稳定性对工艺的影响至关重要,鉴于此,考察了Cu/MS催化剂催化活性与反应时间的关系,结果如图1所示。由图1可知,当反应进行到70 h后,Cu/MS催化剂的DMO转化率和EG选择性开始明显下降,催化剂失活严重。为了进一步改善Cu/MS催化剂的稳定性并抑制难分离副产物1,2-BDO的生成,通过超声辅助浸渍法在Cu/MS催化剂中引入了弱酸性的氧化硼。研究发现,与Cu/MS催化剂相比,xB-Cu/MS系列催化剂的EG选择性明显更高,且无中间加氢产物乙醇酸甲酯(MG)的生成。随着硼负载量的增加,EG选择性呈火山型变化趋势,其中3B-Cu/MS催化剂的EG选择性高达98.24%且无副产物1,2-BDO生成。此外,与未添加硼物种的Cu/MS催化剂相比,3B-Cu/MS催化剂的稳定性大幅提升(>200 h)。弱酸性氧化硼的引入不仅可以改善Cu/MS催化剂的稳定性,还可以抑制难分离副产物1,2-BDO的生成。为了探明弱酸性氧化硼对Cu/MS催化剂结构和表面性质的影响及其与加氢性能间的关系,对xB-Cu/MS系列催化剂进行了系统表征。

图1 Cu/MS和3B-Cu/MS催化剂的催化性能与反应时间的关系Fig.1 Relationship between catalytic performance and reaction time of Cu/MS and 3B-Cu/MS catalysts
2.2 Cu/MS和xB-Cu/MS催化剂的物理结构
采用N2物理吸附仪对Cu/MS和xB-Cu/MS催化剂进行了孔结构表征,结果如图2所示,织构性质如表2所示。根据IUPAC分类,图2中Cu/MS和xB-Cu/MS催化剂的N2吸/脱附曲线均呈现Langmuir IV型等温线,迟滞环为H3型,是典型的平板狭窄型介孔结构。结合表2可知,添加硼物种后,xB-Cu/MS催化剂的比表面积、孔容和孔径均变化不大,说明硼物种的添加对Cu/MS催化剂的孔道结构无显著影响。与MS载体相比,Cu/MS催化剂的织构性质发生较大变化,比表面积、孔容和孔径均有所减小。结合2.3中XRD表征可知,Cu/MS催化剂中出现了层状硅酸铜物相,这可能是由于铜物种和MS载体在催化剂制备过程中发生作用生成层状硅酸铜,进而改变了Cu/MS催化剂的织构性质[21,22]。

图2 Cu/MS和xB-Cu/MS催化剂的N2吸/脱附曲线Fig.2 N2 isothermal adsorption/desorption curves of Cu/MS and xB-Cu/MS catalysts

表2 MS载体、Cu/MS及xB-Cu/MS催化剂的物化参数Table 2 Physicochemical parameters of support and catalysts
2.3 Cu/MS和xB-Cu/MS催化剂的物相结构
为了研究硼物种对催化剂中物相存在状态的影响,采用XRD表征手段对还原前后的催化剂进行分析,结果如图3所示。

图3 Cu/MS和xB-Cu/MS催化剂的XRD谱图:(a)原催化剂,(b)还原后Fig.3 XRD patterns of Cu/MS and xB-Cu/MS catalysts:(a)original,(b)reduced
由图3(a)可知,未还原催化剂样品均在2θ=22°处出现宽化且弥散的衍射峰,该衍射峰归属于无定形SiO2(JCPDS 29-0085)。值得注意的是,所有样品在2θ=31.0°、34.8°、57.2°和63.3°处出现微弱的层状硅酸铜的衍射峰(JCPDS 00-003-0219)[23]。该现象说明,负载的铜物种和MS载体在催化剂制备过程中相互作用生成了层状硅酸铜,且硼物种的引入未影响层状硅酸铜物相的生成。层状硅酸铜物相还原后可以转变为高分散的活性铜物种,有利于提高催化剂的催化活性[23]。此外,xB-Cu/MS催化剂的XRD图谱中未出现任何含硼物种的特征衍射峰,说明硼在催化剂中高度分散。
由图3(b)可知,还原后所有样品在2θ=22°处的无定形SiO2的衍射峰仍然存在,而层状硅酸铜物相的衍射峰消失。此外,所有样品在2θ=36.8°和2θ=43.3°处均出现了明显的衍射峰(JCPDS 05-0667和JCPDS 04-0836),分别归属于Cu+和Cu0。在还原过程中层状硅酸铜转化为高分散的Cu+物种,同时高分散的氧化铜物种转变为高分散的Cu0物种[24]。值得注意的是,随着硼负载量的增加,还原后xB-Cu/MS催化剂中铜物种的特征衍射峰强度明显减弱,说明添加的硼物种通过其与表面铜物种的相互作用提高了催化剂中活性铜物种的分散度。
2.4 Cu/MS和xB-Cu/MS催化剂的形貌结构
采用TEM对MS载体和还原前后的催化剂形貌结构进行表征,表征结果如图4、图5所示。由图4可知,MS载体为均匀介孔结构,而Cu/MS和xB-Cu/MS催化剂中存在大量的晶须结构,进一步证实催化剂制备过程中铜和MS载体相互作用生成了层状硅酸铜物相[25,26]。由图5可知,还原后的铜纳米颗粒在Cu/MS和xB-Cu/MS催化剂上均匀分布。随着硼负载量的增加,铜颗粒尺寸逐渐减小,说明硼物种的添加有利于催化剂中活性铜物种的分散,该结果和XRD表征结果一致。高分散活性铜物种的存在,可以暴露更多的活性位,从而有利于DMO加氢反应的进行。

图4 MS载体及Cu/MS和xB-Cu/MS催化剂的TEM图Fig.4 TEM images of MS support and Cu/MS and xBCu/MS catalysts

图5 还原后Cu/MS和xB-Cu/MS催化剂的TEM图Fig.5 TEM images of reduced Cu/MS and xB-Cu/MS catalysts
2.5 Cu/MS和xB-Cu/MS催化剂的还原性能
采用程序升温还原表征手段(H2-TPR)研究硼物种对催化剂还原性能的影响,表征结果如图6所示。由图6可知,Cu/MS催化剂在244℃左右有一个相对对称的氢气还原峰。研究发现,层状硅酸铜对Cu+和CuO对Cu0具有相同的还原温度,均在237℃左右[27,28]。氧化硼比MS载体更易接受电子,硼物种的引入使得催化剂表面铜物种获得电子的能力变弱,从而导致催化剂中铜物种更难还原,表现为H2还原温度升高[29]。据此,图6中Cu/MS催化剂在244℃左右的还原峰可归为高分散CuO还原为Cu0和层状硅酸铜还原为Cu+的重叠峰;随着硼负载量的增加,xB-Cu/MS催化剂的还原峰位置逐渐向高温方向移动。
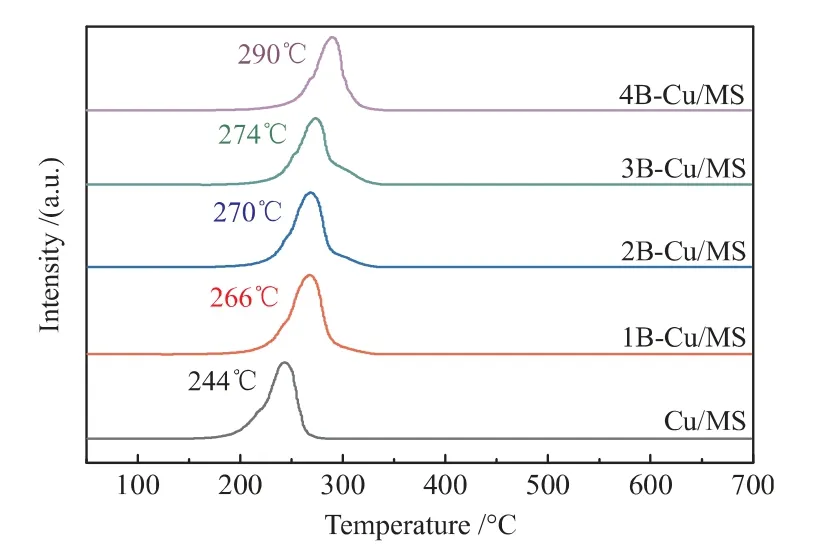
图6 Cu/MS和xB-Cu/MS催化剂的H2-TPR图Fig.6 H2-TPR patterns of Cu/MS and xB-Cu/MS catalysts
2.6 Cu/MS和xB-Cu/MS催化剂的表面酸性
NH3程序升温脱附技术作为一种表征分子筛和负载型金属催化剂等固体催化剂表面酸性质的有效手段得到广泛应用[30]。为了获得硼物种对催化剂表面酸性的影响,将系列硼改性铜基催化剂进行了NH3程序升温脱附表征,结果如图7所示。
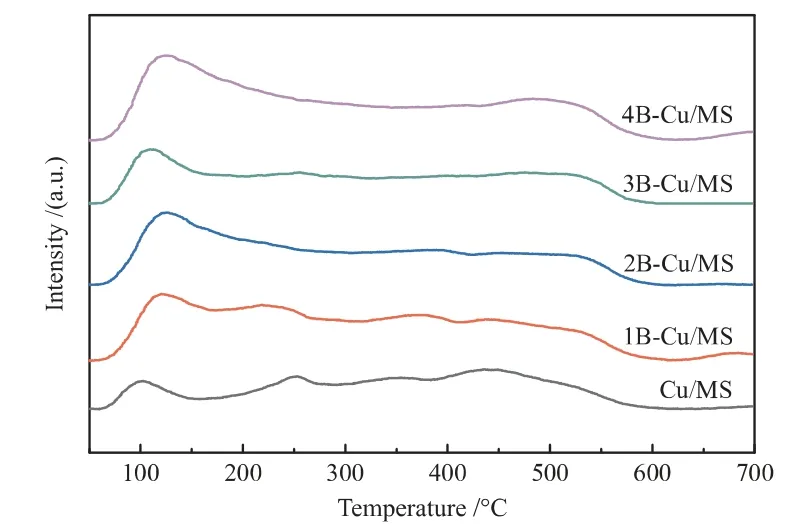
图7 Cu/MS和xB-Cu/MS催化剂的NH3-TPD谱图Fig.7 NH3-TPD patterns of Cu/MS and xB-Cu/MS catalysts
由图7可知,与Cu/MS相比,xB-Cu/MS催化剂的表面弱酸量均有所增加,说明硼物种的添加对催化剂表面弱酸性位点的数量有显著影响[29]。随着硼负载量的增加,xB-Cu/MS催化剂的表面弱酸量呈现先增加后减小再达最大的趋势,其中3B-Cu/MS催化剂的弱酸量最小。此外,3B-Cu/MS催化剂的弱酸峰位置向低温方向移动,说明该催化剂的酸强度和酸量均变小。DMO加氢催化剂的酸碱性对产物分布有较大影响。催化剂中酸性位会导致EG分子间脱水产生EtOH(HOCH2CH2OH+H2→CH3CH2OH+H2O),而碱性位则会通过EG与EtOH之间的Guerbet反应促进1,2-BDO的生成(HOCH2CH2OH+CH3CH2OH→HOCH2CH(CH2CH3)OH+H2O)[20,31]。因此,只有具有适量弱酸性位的催化剂,才能实现抑制1,2-BDO生成的同时,不促进EG脱水生成EtOH。结合表1中Cu/MS及xB-Cu/MS催化剂的活性可知,弱酸性氧化硼的引入,将DMO加氢反应的EG选择性由94.13%(Cu/MS)提高至98.24%(3B-Cu/MS),同时难分离副产物1,2-BDO选择性由0.63%下降至0.00%。由于3BCu/MS的弱酸性位数量和强度低于1B-Cu/MS、2BCu/MS和4B-Cu/MS催化剂,因此该催化剂的EtOH选择性明显较低。该结果表明,适宜硼物种的添加不仅有利于抑制碱性位上1,2-BDO的生成,而且有利于提高目标产物EG的选择性。
2.7 Cu/MS和xB-Cu/MS催化剂的组分和价态
还原后Cu/MS和xB-Cu/MS催化剂的XPS谱图如图8所示。由图8(a)、图8(c)可知,还原后的xB-Cu/MS催化剂均出现了B 1s峰,表明硼物种已成功添加进催化剂中,且随着硼添加量的增加,B 1s的峰逐渐增强。在Cu 2p图中可以看到,还原后的催化剂只出现932.9 eV和952.7 eV两个特征峰,分别归属于Cu 2p3/2和Cu 2p1/2[29]。此外,图中并未出现Cu2+的卫星峰,说明还原后的催化剂中Cu2+已经被完全还原为Cu0或Cu+[32]。由于Cu0和Cu+的结合能非常接近,通过Cu 2p谱图已经无法区分,因此通过Cu LMM谱(图8(d))对Cu0和Cu+进行区分[33]。在Cu LMM谱图中,所有催化剂均存在一个宽泛而不对称的峰,说明还原后的催化剂表面同时存在Cu0和Cu+[34]。为更准确得到Cu0和Cu+的相对含量,对俄歇电子峰进行了分峰处理,其中916.2 eV和913.1 eV处的峰分别归属为Cu0和Cu+[32]。通过拟合结果可以看到,硼物种的添加影响了催化剂表面Cu0和Cu+的分布情况(见表2)。随着硼负载量的增加,Cu+/(Cu++Cu0)的值呈现先增加后降低的火山型趋势。氧化硼作为一种相对亲电物质,在铜物种与氧化硼相互作用时使得铜物种表面具有了偏正电的电荷,从而提高了催化剂中的Cu+占比[22,35]。结合相关表征结果可以发现,适量硼物种的添加对催化剂的物化性质有以下三点影响:(1)有利于活性铜物种的分散,进而改善催化剂的催化加氢活性;(2)降低Cu+物种的还原性且维持适宜的Cu+/(Cu++Cu0)比,可提高催化剂的EG选择性和催化稳定性;(3)提高催化剂表面的弱酸强度和密度,从而有利于抑制副产物1,2-BDO的生成。因此,3B-Cu/MS催化剂不仅具有较高的加氢活性,而且可以抑制副产物1,2-BDO的生成,从而表现出优异的EG选择性和稳定性。

图8 还原后Cu/MS和xB-Cu/MS催化剂的XPS谱图Fig.8 XPS spectra of Cu/MS and xB-Cu/MS catalysts after reduction
3 结论
本文通过尿素协助沉淀结合后浸渍法制备了一系列硼物种改性xB-Cu/MS催化剂。研究表明,适量硼物种引入可提高催化剂表面的弱酸性,从源头上抑制了在碱性位上通过Guerbet反应生成的难分离副产物1,2-BDO。此外,适量酸性氧化硼的引入还可以通过与表面铜物种之间的相互作用提高活性铜物种的分散度和稳定性、维持适宜的Cu+/(Cu++Cu0)比,从而使xB-Cu/MS系列催化剂表现出优异的DMO加氢活性。在180℃、2.5 MPa、质量空速0.4 h-1和n(H2)/n(DMO)为100的反应条件下,3B-Cu/MS催化剂的DMO转化率达100%、EG选择性达98.24%、稳定性达200 h且无副产物1,2-BDO生成。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:36
保鲜与加工(2021年1期)2021-02-06 06:43:22
重型机械(2019年3期)2019-08-27 00:58:44
材料科学与工程学报(2016年2期)2017-01-15 13:34:24
广东饲料(2016年5期)2016-12-01 03:43:22
焊接(2016年9期)2016-02-27 13:05:22
中国资源综合利用(2016年12期)2016-01-22 02:02:26
色谱(2015年6期)2015-12-26 01:57:36
新疆钢铁(2015年2期)2015-11-07 03:27:52
应用化工(2014年1期)2014-08-16 13:34:08
