
不同产地蒙紫草中左旋紫草素和β,β′-二甲基丙烯酰阿卡宁含量测定及质量标准研究*
2021-09-12 05:23吕晓洁刘德旺程丹丹那生桑
中国药业 2021年17期
吕晓洁,渠 弼,刘德旺,程丹丹,那生桑
(1.内蒙古医科大学药学院,内蒙古 呼和浩特010110;2.内蒙古医科大学蒙医药学院,内蒙古 呼和浩特010110)
蒙紫草为紫草科植物疏花软紫草Arnebia szechenyiKanitz.的干燥根,蒙文植物名塔日漠格-希日-伯日漠格。据《内蒙古植物药志(第二卷)》[1]《内蒙古植物志(第四卷)》[2]记载和蒙医用药情况考证,属蒙医自采自用的常用药材。本课题研究的药材为新起草品种,紫草收载于2020年版《中国药典(一部)》[3],为紫草科植物新疆紫草Arnebia euchroma(Royle)Johnst.或内蒙紫草Arnebia guttataBunge的干燥根,二者来源不同。本品药材蒙名和中文名根据《中国蒙药名词规范(2016年版)》紫草名前冠“蒙”而成,植物属名加药用部位自拟拼写了药材品种拉丁名,根据《内蒙古蒙药炮制规范(2015年版)》命名方法确定品名,与药材名一致。疏花软紫草为砾石生旱生植物,生于石质山坡及山坡沟地,见于巴彦淖尔市、阿拉善市、贺兰山等地。蒙紫草根部入药,性凉,味甘、微苦,有清热、凉血、止血、透疹功效。该药虽入味多种蒙医药传统复方制剂,但现代研究甚少,与其他属紫草混用严重。本研究中参考2020年版《中国药典(一部)》紫草项下规定,从药材的性状、显微特征、水分、总灰分、酸不溶性灰分、浸出物、含量测定等方面进行研究,建立其质量标准,为蒙紫草的进一步应用提供了科学依据。现报道如下。
1 仪器与试药
1.1 仪器
U3000型高效液相色谱仪(美国Thermo公司),包括四元泵、在线脱气机、自动进样器、二极管阵列(DAD)检测器、Chromeleon 7色谱工作站;AL204型电子天平(瑞士梅特勒-托利多仪器有限公司,精度为十万分之一);TU-1901型紫外分光光度计(北京普析通用仪器有限责任公司);SX2-4-10型马弗炉(天津市中环实验电炉有限公司);SB5-12DTD型超声波清洗机(宁波新芝生物科技股份有限公司,功率为500 W,频率为40 kHz)。
1.2 试药
左旋紫草素对照品(批号为110769-200506,含量为98%),β,β′-二甲基丙烯酰阿卡宁对照品(批号为111689-201504,含量为98%),均购自中国食品药品检定研究院);GF254薄层板(青岛海洋化工有限公司);甲醇、乙腈均为色谱纯(美国Fisher公司);其他试剂均为分析纯,水为超纯水。蒙紫草药材信息见表1。采摘地点A,内蒙古自治区巴彦淖尔市乌特拉前旗;采摘地点B,内蒙古自治区巴彦淖尔市乌特拉后旗;采摘地点C,内蒙古自治区乌海拉僧庙;采摘地点D,内蒙古自治区乌海老石旦洗煤厂;采摘地点E,内蒙古自治区乌海乌巴公路41 km;采摘地点F,内蒙古自治区阿拉善右旗阿贵庙。经内蒙古医科大学渠弼教授鉴定为紫草科植物疏花软紫草Arnebia szechenyiKanitz.的干燥根。

表1 蒙紫草药材信息Tab.1 Information of Arnebiae Szechenyi Radix
2 方法与结果
2.1 药材性状
本品呈圆锥形或圆柱形,稍扭曲,长6~15 cm,直径0.5~3.0 cm。根头部略粗大,顶端有残茎多个,被短硬毛;表面紫红色或灰紫色,皮部常数层相叠,易剥离。质硬而脆,易折断,断面较整齐,皮部紫红色,木部黄白色;气特异,味微甘、微涩。
2.2 药材鉴别
2.2.1 显微鉴别
横切面:木栓层1至多层,将皮层及韧皮部分隔呈层状,外侧皮层及韧皮部多颓废呈裂隙状。内侧皮层狭窄,由2~3层薄壁细胞组成,细胞多切向延长。韧皮部宽广,射线细胞多列放射状,韧皮束内侧细胞常木栓化。束间形成层明显,木质部放射状,外侧导管多径向排列,老根中心具木栓层环,环内组织除导管外多颓废呈裂隙状。木栓细胞和部分薄壁细胞含紫色素。详见图1。

图1 蒙紫草药材横切面特征(×40)A.Cross sectional characteristics of Arnebiae Szechenyi Radix B-C.Cork layer and cortex D-E.Phloem,cambium and xylem F.Decadent xylem in the center of old root G.Interfascicular cambiumFig.1 Cross section characteristics of Arnebiae Szechenyi Radix(×40)
粉末:呈深紫红色;非腺毛单细胞,直径13~36μm,直立或稍弯曲,壁光滑或具壁疣,部分胞腔内含紫棕色色素;栓化细胞红棕色,表面观呈多角形或圆多角形,含紫红色色素;薄壁细胞较多,淡棕色或无色,大多充满紫红色色素;导管主为网纹导管,少有具缘纹孔导管,直径9~60 μm。纤维纹孔明显,直径7~15 μm。详见图2。
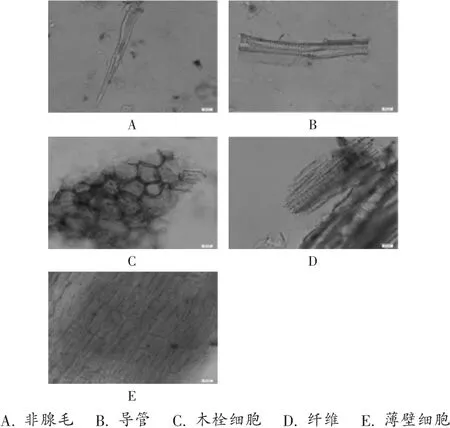
图2 蒙紫草药材粉末特征(×400)A.Non-glandular hair B.Vessels C.Cork cells D.Fiber E.Parenchyma cellsFig.2 Powder characteristics of Arnebiae Szechenyi Radix(×400)
2.2.2 薄层鉴别
取本品粉末0.5 g(过3号筛),精密称定,加石油醚(60~90℃)20 mL,超声处理(功率为500 W,频率为40 kHz)20 min,滤过,滤液浓缩至1 mL,作为供试品溶液。取左旋紫草素和β,β′-二甲基丙烯酰阿卡宁对照品适量,用乙醇配成质量浓度为1 mg/mL的对照品溶液。照2020年版《中国药典(四部)》通则0502薄层色谱法试验,吸取上述溶液各4 μL,分别点于同一硅胶板上,以环己烷-甲苯-乙酸乙酯-甲酸(5∶5∶0.5∶0.1,V/V/V/V)为展开剂,展开,取出,晾干。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同紫红色斑点,再喷以10%氢氧化钾甲醇溶液,斑点变为蓝色,且斑点均分离清晰。详见图3。
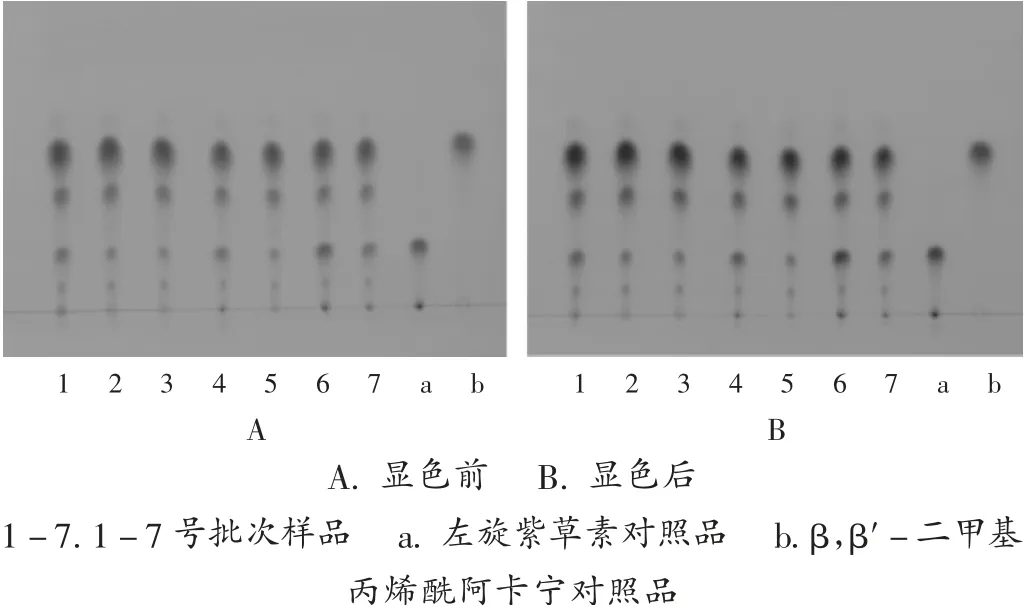
图3 蒙紫草薄层色谱图A.before color development B.after color development1-7.Samples of batches 1-7a.L-shikonin reference substance b.β,β′-dimethylacrylalkannin reference substanceFig.3 TLC chromatograms of Arnebiae Szechenyi Radix
2.3 水分、灰分、浸出物检查
本品属根类药材,根皮失水后较脆且薄,根据前期研究,有效成分中含有水溶性物质,不适合用水清洗,处理和采集过程中易带入泥土等杂质而影响药材质量,故参考2020年版《中国药典(四部)》通则2302检查其总灰分和酸不溶性灰分。此外,药材含水量反映其干燥程度,影响有效成分的稳定性和批量加料时的准确性,合适的水分含量可防止储存过程中的虫害与霉变,避免有效成分分解、酶解变质等,故进行水分检查(通则0832第二法)。同时,参考通则2201水溶性浸出物测定法的冷浸法、醇溶性浸出物测定法,对收集药材的水溶性和醇溶性浸出物进行了含量测定。结果见表2。考虑试验主要在秋冬干燥季节进行,故规定蒙紫草水分含量不得过12.00%;其余项目的含量,结合具体情况,初步制订质量标准为:总灰分含量不得过8.00%,酸不溶性灰分含量不得过2.00%,水溶性浸出物含量不得低于30.00%,醇溶性浸出物含量不得低于10.00%。

表2 样品水分、总灰分、酸不溶性灰分、水溶性浸出物及醇溶性浸出物平均含量测定结果(%,n=3)Tab.2 Results of content determination of water,total ash,acid insoluble ash,water-soluble extract and alcohol soluble extract in the sample(%,n=3)
2.4 左旋紫草素含量测定
2.4.1 测定波长选择
参照2020年版《中国药典(四部)》通则0401紫外分光光度法,将对照品溶液进行全波长扫描,在516 nm波长处有最大吸收,供试品在相应波长也有最大吸收,阴性对照无干扰,故选择516 nm为紫外测定波长。
2.4.2 提取方法考察
取粉碎后过3号筛的蒙紫草药材0.5 g,精密称定,共3份,分别加入石油醚(60~90℃)和乙醇100 mL,分别浸泡4,5,6 h,不时振摇,滤过。精密量取续滤液5 mL,转移至25 mL容量瓶中,加溶液至刻度,摇匀,于516 nm波长处测量吸光度,每份重复测定3次。结果同种提取溶剂提取时间越长,提取量越大,乙醇作提取溶剂时,含量增加更快。综合考虑石油醚和乙醇的提取效果,并考虑时间、提取溶剂的毒性及溶剂的成本问题,参考同属药材紫草2020年版《中国药典(一部)》中的提取方法,结合药物稳定性综合考虑,选择提取溶剂为乙醇,提取时间为5 h。
2.4.3 溶液制备方法
取粉碎后过3号筛的蒙紫草药材0.5 g,精密称定,加乙醇100 mL,浸泡5 h,不时振摇,滤过,精密量取续滤液5 mL,转移至25 mL容量瓶中,加溶液至刻度,摇匀,即得供试品溶液。取左旋紫草素对照品,精密称定,加乙醇制成质量浓度为200 μg/mL的对照品溶液。
2.4.4 方法学考察
线性关系考察:将对照品溶液逐级稀释,于516 nm波长处进样测定2次,测量吸光度,取平均值,以左旋紫草素质量浓度(X,μg/mL)为横坐标、吸光度(Y)为纵坐标进行线性回归,得线性回归方程Y=0.024 6X+0.010 3,R2=0.999 9(n=2)。结果表明,左旋紫草素质量浓度在5~50 μg/mL范围内与吸光度线性关系良好。
精密度试验:依法制备供试品溶液,于516 nm波长下连续进样测定6次。结果左旋紫草素吸光度的RSD为0.16%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(批号为181011)样品,依法制备供试品溶液,于室温下放置0,1,2,4,6,8 h时测量吸光度。结果左旋紫草素吸光度的RSD为0.58%(n=6),表明供试品溶液在室温下放置8 h稳定。
重复性试验:取同一批(批号为181011)蒙紫草粉末,精密称定,依法制备供试品溶液,于516 nm波长下进样测定6次,记录吸光度值,并计算含量。结果左旋紫草素的平均含量为2.60%,RSD为1.72%(n=6),表明方法重复性良好。
加样回收试验:取同一批(批号为181011)已知含量的蒙紫草粉末0.1 g,精密称定,共9份,依法制备供试品溶液,按50%,100%,150%低、中、高水平加入对照品,测定含量,并计算回收率。结果见表3。
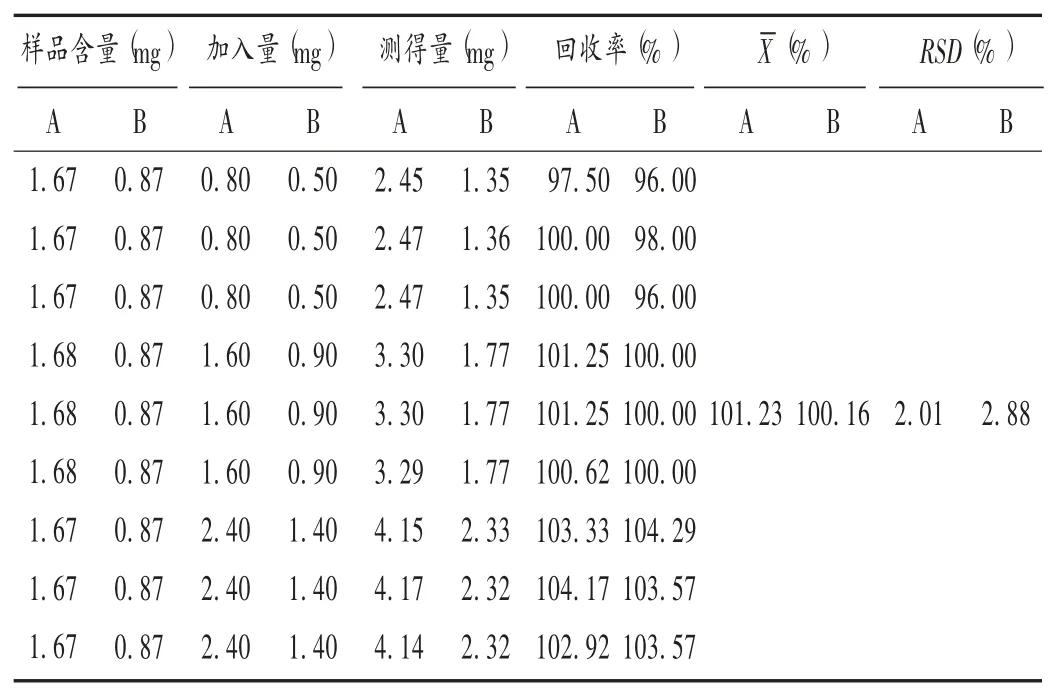
表3 加样回收试验结果(n=9)Tab.3 Results of the recovery test(n=9)
2.4.5 样品含量测定
取1-7号批次样品,依法制备供试品溶液,分别于516 nm波长处进样测定3次,并计算含量。结果样品中左旋紫草素(以干燥品计)的含量介于1.40%~4.09%,平均含量为2.49%。考虑到不同地点、不同时间采集的药材含量差异较大,故拟订本品羟基蒽醌总色素(以左旋紫草素计)不得少于1.00%。
2.5 β,β′-二甲基丙烯酰阿卡宁含量测定
2.5.1 提取方法考察
取蒙紫草药材粉末0.5 g(过4号筛),精密称定,共4份,分别加入石油醚(60~90℃)和乙醇25 mL,分别超声(功率为250 W,频率为33 kHz)处理30 min和45 min,放冷,称定质量,加相应溶剂补足减失的质量,摇匀,滤过。精密量取续滤液5 mL,蒸干,残渣加流动相溶解,转移至5 mL容量瓶中,加流动相至刻度,摇匀,滤过,取续滤液,分别进样测定3次,记录峰面积,并计算含量。结果石油醚超声30 min提取时含量最高。
2.5.2 色谱条件与系统适用性试验
色谱柱:Thermo Hypersil C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.05%甲酸水溶液(70∶30,V/V);流速:1.0 mL/min;检测波长:275 nm;柱温:25℃;进样量:10 μL。理论板数按β,β′-二甲基丙烯酰阿卡宁峰计应不低于5 000。在此色谱条件下,供试品溶液色谱中,在与对照品溶液色谱相应位置处有色谱峰出现,且分离度良好,阴性对照无干扰,表明专属性良好。详见图4。
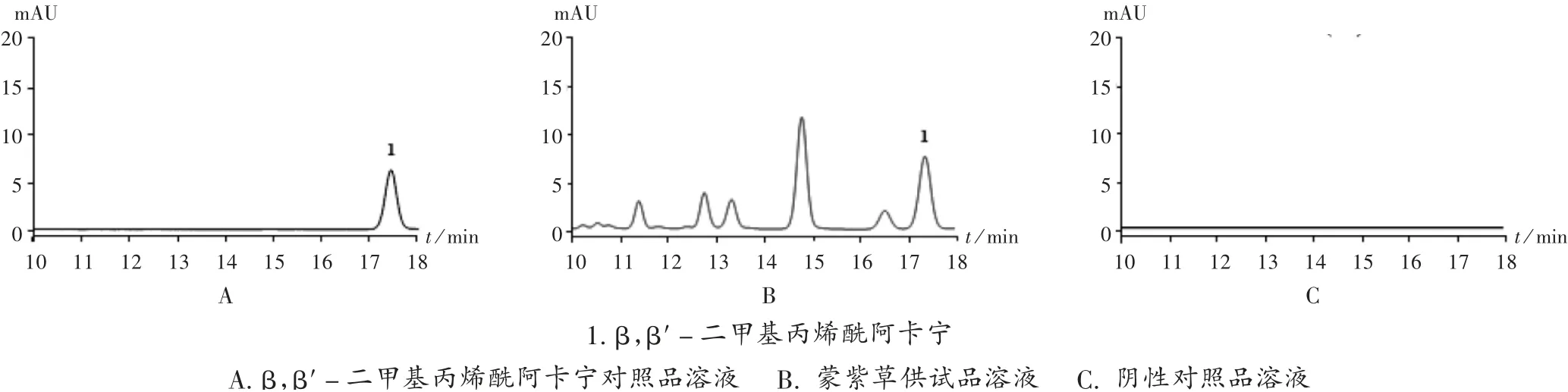
图4 β,β′-二甲基丙烯酰阿卡宁高效液相色谱图1.β,β′-dimethylacrylalkanninA.β,β′-dimethylacrylalkannin reference solution B.Arnebiae Szechenyi Radix test solution C.Negative reference solutionFig.4 HPLC chromatograms of β,β′-dimethylacrylalkannin
2.5.3 溶液制备
取过4号筛的各批次药材0.5 g,精密称定,加入石油醚(60~90℃)25 mL,称定质量,超声30 min,放冷,补足减失的质量,摇匀,滤过,精密量取续滤液10 mL,蒸干,残渣加流动相溶解,转移至10 mL容量瓶中,加流动相至刻度,摇匀,滤过,取续滤液,即得供试品溶液。取β,β′-二甲基丙烯酰阿卡宁对照品,精密称定,加无水乙醇配制成质量浓度为100 μg/mL的对照品溶液。
2.5.4 方法学考察
线性关系考察:依法制备对照品溶液,逐级稀释,按拟订色谱条件进样测定2次,取平均值,以峰面积(Y)为纵坐标、质量浓度(X,μg/mL)为横坐标进行线性回归,得回归方程Y=0.208 9X+0.118 4,R2=0.999 6(n=2)。结果表明,β,β′-二甲基丙烯酰阿卡宁质量浓度在10~200 μg/mL范围内与峰面积线性关系良好。
精密度试验:取同一批(批号为181011)样品,依法制备的供试品溶液,按拟订色谱条件连续进样10 μL,测定6次。结果峰面积的RSD为0.21%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(批号为181011)样品,依法制备供试品溶液,分别于室温下放置0,1,2,4,6,8,10 h时按拟订色谱条件进样测定。结果峰面积的RSD为0.51%(n=7),表明供试品溶液室温下放置10 h内稳定。
重复性试验:取过4号筛的药材粉末6份,依法制备供试品溶液,按拟订色谱条件进样测定,并计算含量。结果含量的RSD为1.31%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的同一批(批号为20181011)蒙紫草粉末(过4号筛)0.5 g,精密称定,共9份,依法制备供试品溶液,按50%,100%,150%低、中、高水平加入对照品,按拟订色谱条件进样测定,计算加样回收率。结果见表3。
2.5.5 样品含量测定
取1-7号批次样品,依法制备供试品溶液,按拟订色谱条件进样测定,并计算含量。结果样品中β,β′-二甲基丙烯酰阿卡宁(按干燥品计)含量介于0.20%~0.33%,平均含量为0.26%。考虑到不同地点药材含量的差异及阿卡宁的稳定性,拟订本品含β,β′-二甲基丙烯酰阿卡宁不得少于0.15%。
2.6 蒙紫草质量标准拟订
综合考虑成分特征、对照品来源、测量便利,参考同科植物的药典收载情况[3]、文献[4-7]报道及预试验结果,最终采用紫外分光光度法测定羟基蒽醌总色素(以左旋紫草素计)的含量,采用高效液相色谱法测定β,β′-二甲基丙烯酰阿卡宁的含量。虽然该药材在内蒙古地区多有分布,但成分含量差异很大,入药效果也有差异。表4列出了8-12号批次药材的含量测定及检查各项数据。综合分析12批次药材中左旋紫草素和β,β′-二甲基丙烯酰阿卡宁的含量,发现1-7号批次药材中这2种成分的含量较高。参考文献[1,5-8],认为紫草中萘醌类成分为其主要药效成分,参考2020年版《中国药典(一部)》中同属植物紫草项下的含量测定项,根据1-7号批次药材的产地和采收时间初步拟订了蒙紫草的质量标准。

表4 8-12号批次样品中含量测定及检查项测定结果(%,n=3)Tab.4 Source of Arnebiae Szechenyi Radix samples of batches 8-12,the results of content determination of L-shikonin and β,β′-dimethylacrylalkannin in the samples,and the results of inspection items(%,n=3)
3 讨论
同科的紫草类植物中,萘醌类是其主要有效成分[4],主要含紫草素和阿卡宁类[9],用于治疗炎症、烧伤、麻疹等疾病,可促进伤口愈合[5-7]。紫草素诱导调节氧化应激反应、线粒体功能和细胞骨架形成的信号通路,可作为开发新型抗癌药的来源化合物[10],与临床已有抗癌药物联用,可协同抑制肿瘤细胞生长[11]。
从采摘地点来看,乌海拉僧庙、乌海老石旦洗煤厂、乌海乌巴公路41 km处及阿拉善右旗阿贵庙收集的样品中,β,β′-二甲基丙烯酰阿卡宁和左旋紫草素的含量均远低于巴彦淖尔市乌拉特前旗、后旗收集的样品。从采摘时间分析,11月收集的样品含量普遍低于10月收集的样品。综合分析建议,该药材的最佳采收时间应在初秋,采收地点首选巴彦淖尔市乌拉特前旗、后旗附近。
药材的质量标准对于其临床应用、新药研发、传统药物现代化、中蒙药资源开发,乃至整个中蒙医药产业的健康、可持续发展均有重要意义。本课题组采集的药材是新起草品种,采摘地点位于内蒙古的主要紫草产区,检测方法为目前普遍认可的药物质量标准测定方法,制订的质量标准较完善,可为该药材的质量控制与药品品质鉴定提供科学依据,对于该药材的人工种植、资源扩大有重要意义。后续需进一步对药材的采收时间、药理、药效及不同种属之间药材的差异进行更深入的研究。
猜你喜欢
现代工业经济和信息化(2022年7期)2022-09-02
西部散文选刊(2022年1期)2022-02-03
电脑报(2021年35期)2021-09-16
天然产物研究与开发(2020年4期)2020-06-02
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
布达拉(2019年3期)2019-06-11
食品界(2017年4期)2017-05-17
中国中药杂志(2017年8期)2017-05-11
百科知识(2016年18期)2016-10-28
