
土壤硒的存在特征及分析测试技术研究进展
2021-09-09 06:57伊芹程皝尚文郁
岩矿测试 2021年4期
伊芹, 程皝, 尚文郁
(1.国家地质实验测试中心, 北京 100037;2.北京航天威科环保科技有限公司, 北京 100071)
硒作为重要的生命健康微量元素之一,近年来引起社会广泛关注。土壤硒的全球异质性分布是造成各种病害(因硒过剩/缺乏导致)和环境问题的主要原因[1]。准确分析土壤硒含量是促进生态地球化学、环境科学等相关方面研究的基本前提。
当前,应用于土壤硒的分析技术主要包括光谱技术和质谱技术,不同分析方法的适用范围和技术优势不同[2-4]。充分了解样品的来源特征有助于针对性地选择分析方法,合理匹配高基体相似度标准物质进行质量监控能够有效地提高分析质量。全球大部分地区的硒含量较低,土壤硒存在形式和分布特征受多种因素共同影响,易发生变化,人类活动加剧了其变化速度[5-7]。受限于分析技术的发展和标准物质研制的滞后,当前仍存在部分硒物种无法定性定量分析、土壤硒迁移转化机理尚不完善的问题[8-9]。上述问题的存在对土壤硒分析技术的发展和标准物质研制提出了挑战,反过来也制约了硒分析测试质量的提高。发展高精准度的痕量、超痕量分析技术,及时扩展土壤硒标准物质的基体类型和含量范围,成为当前土壤硒分析领域的硬性要求。
本文从自然和人为活动两方面分析了土壤硒的来源和土壤硒的主要存在形式;综述了光谱和质谱技术在土壤硒分析领域的应用,比较了不同分析方法的检出限、精密度和线性范围。重点论述了碰撞反应池技术在去除质谱干扰方面的研究进展,以期为分析方法的选择提供参考。同时阐述了国内外土壤硒标准物质研制现状,指出及时扩展土壤硒标准物质的基体类型和含量范围的必要性。
1 土壤硒来源及土壤硒存在形式
在世界范围内,土壤硒的分布不均匀,具有显著的空间异质性。除典型高硒地区外,土壤硒含量通常在0.01~2.0mg/kg范围内,平均值一般为0.4mg/kg[9]。中国大部分地区的土壤贫硒,土壤硒含量介于0.119~0.344mg/kg[10],平均值为0.239mg/kg[11];在典型高硒地区,土壤硒含量可达346~2018mg/kg[12]。
1.1 土壤硒的来源
土壤硒含量的增减是自然过程和人类活动共同作用的结果。成土母质是土壤硒的主要地质来源,人类活动对硒循环的影响日益加重。图1描述了自然界硒的循环活动。
1.1.1自然过程
火山喷发和成土母岩风化淋滤是增加土壤硒含量的主要自然过程。火山喷发时,熔岩流、火山灰流和火山碎屑流直接将含硒物质释放到环境中,并通过大气沉降和水文作用进入土壤;火山灰和岩石也可风化成肥沃的火山土壤。据报道,爱尔兰火山区土壤硒含量高达1200mg/kg[13]。夏威夷火山土壤硒含量为1~20mg/kg[14],明显高于美国其他地区土壤硒含量。但火山土壤不足全球地表面积的0.8%[14]。
成土母质是影响土壤硒含量的主要自然因素。当代全球硒源岩分布与古富有机质沉积海相盆地密切相关,硒含量较高的土壤多来源于前寒武纪富含有机质的碳酸盐岩[15]。由于硒可被黏土矿物强烈吸附,并可被水生生物富集,故富含黏土、有机碳的沉积岩(如泥岩和页岩)含硒量相对较高[16]。黑色页岩是古代富有机质沉积的结果,其含量较高的地区,均有高硒土壤报道。如爱尔兰页岩区[17]、英格兰北部Bowland页岩组的石炭纪黑色页岩区[18]、美国密西西比州巴尼特页岩区和科罗拉多州白垩纪Mancos页岩区[19]。据分析,中国恩施富硒土壤成因也与二叠系地层炭质、硅质、含炭页岩有关[20]。而在潮湿气候下发育的砂质土壤,尤其是灰化土,其黏粒和有机质含量均较低,不易富集硒,通常含硒量较低[21]。
成土母质不是影响土壤硒含量的绝对因素。中国土壤硒含量远高于土壤母岩硒含量,特别是在西北和东南地区。硒在中国三大主要岩石中的含量分别为:变质岩0.070mg/kg、火成岩0.067mg/kg、沉积岩0.047mg/kg。硒在中国不同地区土壤中的含量分别为:西北地区 0.19mg/kg、中部低硒带0.13mg/kg、东南沿海 0.23mg/kg[22]。中国缺硒带的土壤平均硒含量约0.125mg/kg[23],同样高于岩石硒含量。
1.1.2人类活动
土壤硒人为来源主要通过矿山开采、有色金属生产和制造等活动释放[24]。采矿活动中,硒可通过氧化废石和高浓度废水进入下游地表水和土壤中[5]。硒的地球化学性质与硫相似,常以类质同象方式进入硫化物晶格,与硫化物矿物伴生。硒在硫化物矿物中的含量最为丰富,尤其是与砂岩型矿床中的铀矿石、黄铁矿和沉积岩中的类金属矿床有关的硫化物矿物。例如,方铅矿的硒含量通常为0~15mg/kg;毒砂的硒含量为42~57mg/kg);磁黄铁矿的硒含量为5~63mg/kg;马卡锡石的硒含量为0~11mg/kg;黄铜矿的硒含量为10~50mg/kg;黄铁矿的硒含量为0~50mg/kg;闪锌矿的硒含量为10~50mg/kg,部分高达900mg/kg[25]。
在富含上述矿种的地区,土壤硒含量通常较高[6],如美国布莱克福特流域二叠系磷矿开采造成硒污染,引发牲畜死亡[26]。
下列公式(1)和公式(2)描述了含硒黄铁矿被氧气和硝酸氧化释放硒的过程。
(1)
7N2+2H2O
(2)
此外,农业活动中人为地向土壤中添加含硒肥料、使用硒污染水灌溉、化石燃料的精炼和燃烧等,均能增加土壤硒含量[7]。如印度西北部旁遮普邦和哈里亚纳邦农业区的土壤硒含量高,即归因于该地区富硒灌溉水[27]。化石燃料的精炼和燃烧活动直接向土壤排放含硒物质,也是造成灌溉水硒污染的原因之一。焦化厂附近土壤常表现出明显的硒富集[28]。可以看出,人类活动对土壤硒含量的影响,使得高硒土壤不再仅限于典型地质区域。
1.2 土壤硒的主要存在形式
随着土壤硒研究的深入,已确定土壤硒的具体存在形式影响硒的流动性和生物毒性[29-30]。仅对总硒含量进行分析无法满足研究需求。
土壤硒可以多种化合物形式存在,如无机硒化合物和有机硒化合物,同时以多种价态存在,如Se2-、Se0、Se4+、Se6+。表1汇总了土壤中常被报道的硒化合物。硒的存在形式和含量受环境影响动态发生变化。由于无机硒化合物的毒性强于有机硒化合物(高约40倍)[31],且无机硒化合物能被植物直接吸收利用,早期针对无机硒的分析和研究相对较多。其中,硒酸盐和亚硒酸盐可以离子形式存在于土壤水溶相,或可被土壤有机质、含氧化合物和土壤黏土吸附[32-34],生物利用度和迁移性相对较高,是研究和分析中较常关注的物种。随着研究的深入,有关土壤有机硒的分析逐渐增多,但受限于检测技术的发展和标准物质的匮乏,仍有大量有机硒物种有待确定。

表1 土壤中部分含硒化合物及其存在环境
土壤中硒含量和存在形式受土壤化学环境和物理变量梯度驱动,该驱动力又由同时跨越多个空间和时间尺度的生物和环境过程所控制。具体包括:土壤所属地区的气候、地形、水文地质条件、土壤微生物、植物和人类活动等[13,16,36,39]。鉴于土壤系统的复杂性和多种影响因素的交互性,且受限于分析技术的不足,土壤硒物种迁移转化理论体系尚不完善,其中涉及的定量关系有待阐明[8]。
2 土壤硒形态提取和分析测试技术
2.1 土壤硒形态分步提取技术
土壤硒形态分析是对硒的各种原子和分子组成形式进行定性和定量分析的过程。由于土壤基体的复杂性和土壤硒形态的易变性,直接对上述组分进行提取存在诸多困难。
分步提取技术通过人为选择专性提取剂,模拟硒在土壤地球化学过程中的迁移转化、生物富集等行为,选择性提取与土壤特定组分相结合的硒物种。该技术易于实施,对问题的解释具有一定的针对性和灵活性,是当前土壤硒化学形态分析的替代方案[40-41]。
当前国内外广泛使用的分步提取方案大都是在Tessier等[42]提出的土壤、沉积物样品重金属元素顺序提取法基础上发展起来的,此后研究者多结合具体研究目的,在上述基础上对提取方案进行改进[43]。表2总结了当前较为常用的几种适用于土壤硒形态分析的分步提取方案。
从表2中可以看出,不同研究采用的提取程序各不相同。国内外尚未统一土壤硒分步提取的分级和操作标准,不同研究数据间缺乏可比性。原因是硒提取方案的产生依托于各自研究需求,并随着研究的深入不断改进。在研究初期,水提取态硒被认为是最能反映植物可利用硒的形态,随后研究发现水提取态中常含有可溶性有机硒化物,有机硒化物无法被植物直接吸收利用,证实水提取态难以直接体现生物可利用硒含量,故而不断促进了对方法的改进。如提出以磷酸缓冲溶液、氯化钾、碳酸氢钠等为提取剂的土壤硒有效态提取方案[51],以0.1mol/L氢氧化钠为提取剂的亚硒酸盐和硒酸盐提取方案[52]等。现阶段,鲜有研究针对提取程序设计的合理性开展深入讨论。大多是通过实际应用效果进行检验。
当前各种分步提取研究方法仍存在诸多问题:①提取剂缺乏特异选择性;②提取过程中,硒形态易受提取剂的影响发生吸附和迁移转化。Jessica等[50]将顺序提取后的分析结果与同步辐射技术原位形态分析结果进行对比,结果显示在指定的提取态提取过程中,目标硒物种未被完全提取,表明所选提取剂缺乏特异性。同样地,Dodd等[53]的研究发现了类似问题,经盐酸羟胺提取铁氧化物结合态后,土壤中仍残留有部分铁氧化物,意味着与土壤铁氧化物相互作用的硒物种没有完全被释放出来。
2.2 土壤硒分析测试技术
2.2.1原子光谱分析技术
应用于土壤硒含量分析的光谱技术主要有原子吸收光谱(AAS)和原子荧光光谱(AFS)。二者均需与进样装置结合使用,常见的有电热原子化进样装置(ET)、氢化物发生进样装置(HG)和石墨炉原子化进样装置(GF)。表3总结了若干样品前处理方法与原子光谱技术联用,分析土壤硒含量的应用实例。
(1)氢化物发生-原子荧光光谱法
氢化物发生-原子荧光光谱(HG-AFS)分析技术因具备如下优点,在中国土壤硒含量分析领域应用较为普遍。①硒元素能够以气液分离方式从基体中脱出,较大限度地减少基体干扰。但仍存在干扰问题(主要是过渡金属离子所产生的液相干扰),大部分可通过添加掩蔽剂的方法得到控制,如通过加入铁盐、增加酸度或采用铁氰化钾作掩蔽剂,可消除部分金属离子的干扰;10%硫脲-10%抗坏血酸作为混合改进剂可消除铜、银、镍、铅等金属离子的干扰[64]。②能获得更低的检出限和相对较高的精密度。如表3所示,HG-AFS技术的检出限可达ng/g数量级水平。原因是AFS测量的是背景相对较低的小荧光信号,而不是两个信号之间的差异。③操作相对简便,仪器价格较低。


表3 原子光谱技术分析土壤硒含量应用实例
HG-AFS分析技术的缺点是线性范围较窄。分析硒含量较高的样品时,只能通过稀释样品溶液的方式解决,但大比例稀释通常会引入较大分析误差,国际上亦不认可此类解决方案。造成该问题的原因是:原子荧光强度与试样浓度仅在低浓度范围内成正比;高浓度下测量光路外的原子浓度会影响分析信号,使其检测到的荧光强度为原子化槽的几何形状的复杂函数。该问题的存在从本质上限制了AFS在土壤硒含量分析中的应用。美国国家环境保护局(EPA)并未将其列为硒元素分析的标准方法,国际期刊中关于原子荧光光谱分析硒含量的报道也相对较少。
此外,使用AFS方法直接分析,仅能测量能够被完全分解矿化、并还原为四价硒的硒物种含量。这就要求分析者根据分析需求和样品特点,合理选择前处理方法。如对土壤样品进行分步提取处理,分析水溶态硒含量时,若直接将水提取液经盐酸还原后分析,则其中可能含有的水溶性有机硒化合物无法被盐酸直接还原为无机态四价硒,分析结果仅为水溶态无机硒含量[29]。若要求分析全部水溶态硒含量,则应添加高氯酸加热处理步骤,将其中可能存在的有机硒物种完全矿化分解。而使用高氯酸消解的步骤在当前顺序提取标准方法中未作出说明[50,65]。
(2)原子吸收光谱法
原子吸收光谱法(AAS)的优点是该方法使用瞬态信号,可通过改性剂辅助热解/雾化,经温度控制程序和吹扫气体保持原子蒸汽的保留时间,增强吸收,不存在光路外干扰问题,可分析浓度的线性范围较广。相较AFS法,AAS法的检出限稍高(表3),但通过在样品处理阶段添加萃取富集步骤能够改善该问题[2]。
采用AAS技术分析土壤硒含量存在的主要问题包括进样损失、基体干扰、谱线干扰等[66]。如石墨炉AAS技术分析土壤硒,由于硒是易挥发元素,灰化温度升至400℃时会产生灰化损失;分析结果受样品基体影响较为严重,需加入基体改进剂减小灰化损失。土壤样品中的Pd2+、Cu2+、Ni2+、Co2+等离子对硒的测定有增感效应,产生正干扰;Mo6+、Pt4+等离子对硒的测定有弱化效应,产生负干扰。基体改进剂的加入能够减小样品基体中干扰离子对硒测定的影响。铱-硝酸镁、钯-硝酸镁等作为测定硒的基体改进剂,均能提高硒的灰化温度和分析灵敏度。与此同时,使用常规线源原子吸收光谱仪时,在195.950nm、196.061nm和196.147nm附近存在三条铁谱线,可能会对最灵敏的硒谱线196.026nm产生光谱干扰[59]。该谱线无明显精细结构条带,利用背景校正(塞曼或氘背景校正)可在一定程度上克服基体光谱干扰,但常存在校正过低或过高的问题[56],使用高分辨连续光源能够在一定程度上改善该问题,是该技术的发展趋势[59]。
2.2.2质谱分析技术
电感耦合等离子体质谱技术(ICP-MS)能够实现硒与多种元素的同时测定。美国EPA发布的多种微量元素(包括硒在内)标准方法中,均使用ICP-MS[2]。该方法在土壤硒分析中面临的主要问题是来源于离子源和样品基体的质谱干扰。碰撞/反应池技术(CRC)和同位素稀释质谱(IDMS)的出现,为解决质谱干扰问题提供了有效途径。前者通过物理碰撞或化学反应消除干扰因素,后者通过离子选择设备甄别干扰。
(1)四极杆电感耦合等离子体质谱技术
使用常规四极杆ICP-MS分析土壤硒含量无法达到满意效果,原因是:①硒元素具有较高的电离能(9.75eV),在氩气等离子体中的电离程度相对较低;②存在严重质谱干扰,包括产生于氩气等离子体离子源处的干扰和来自样品基体的干扰。因大部分干扰信号高于硒信号,无法通过线性方程扣除。表4列出了ICP-MS分析土壤硒含量时常见的同质异位素干扰。

表4 质谱分析土壤硒常见同质异位素干扰
从表4中可以看出,使用ICP-MS分析硒的任一种同位素,均会受到质谱干扰。其中,80Se+丰度比最高,检测灵敏度高,但常规四极杆质谱分析模式下,80Se+受40Ar40Ar+等氩基干扰影响较大。土壤中大量共存离子亦对80Se+产生干扰,如氧、氢、钙、钾、硫、镍、锌、溴和稀土元素。
(2)碰撞反应池技术
碰撞反应池(CRC)技术被认为是一种强大的,理论上可实现无干扰测定的技术[67-68],是当前质谱领域的热点技术。通过将碰撞技术(CCT)和动态反应技术(DRC)与ICP-MS联用,提高了ICP-MS分析硒元素的能力。其工作原理是将碰撞/反应气引入封闭反应池,通过碰撞或化学反应,实现同质异位素分离、离子消除、复合分子离子分裂,从而降低干扰,提高灵敏度[69]。目前商品化的CRC系统主要分为四类:①四极杆动态反应池(美国PerkinElmer公司,主流产品DRC-e和DRCⅡ);②六极杆碰撞反应池(美国ThermoFisher公司,CCT);③八极杆碰撞/反应池(美国Agilent公司,ORS);④碰撞反应接口(CRI,美国Varian公司)。不同产品设计理念有所不同,但去干扰原理类似。
碰撞反应池技术应用于土壤硒分析的核心问题是碰撞/反应气的选择和质谱参数的优化,相关研究之间尚未形成统一标准,原因是样品基体差异和不同仪器设计差异。常见的碰撞/反应气有氦气、氩气、氢气、氨气、甲烷、氮气、氧气、一氧化二氮、一氧化氮、二氧化碳等[67-68]。其中常用于土壤硒分析的有氢气、甲烷、氧气和氦气。屈明华等[70]比较了甲烷和氧气作反应气的应用效果,结果显示在甲烷模式下,1μg/L硒标准溶液信号强度大于4000cps;在氧气模式下,1μg/L硒标准溶液信号强度大于2000cps。Bullock等[66]和Henn等[69]采用氢气作反应气,有效减少了m/z77、78和80处氩基多原子离子的质谱干扰,但无法消除溴、锗对77Se、80Se和82Se的干扰,故仅能通过监测78Se分析煤中硒含量,在优化条件下,仪器和方法的检出限为0.01μg/L。de Feudis等[71]使用具有八极杆碰撞反应系统的ICP-MS(Agilent 7900)分析了土壤中多个提取态硒含量,经与标准物质给定值比较,结果符合要求。Liu等[72]采用微波消解ICP-MS碰撞模式监测82Se,建立了分析土壤硒的方法,检出限为0.1449ng/L,线性范围为1~100μg/L。并分析了土壤形态成分标准物质GBW07445中的硒,RSD为4.4%。
值得关注的是,在质谱分辨率不足的前提下,仅依靠CRC技术无法完全消除硒的质谱干扰,与多重四极杆技术的结合能够较好地解决该问题。林立等[73]分别采用低分辨四极杆质谱常规模式、加氦加氢模式、加氧模式和加氧-双四极杆(SQ-O2)模式对土壤标准物质中的80Se进行分析,结果表明在常规模式下进行土壤硒分析,干扰较多,无法得到满意结果;加氦加氢模式无法消除稀土干扰;在加氢模式下,加重了79BrH+带来的干扰;采用加氧单四极杆模式分析,虽然解决了氯、溴、稀土的干扰问题,又引发了锗、锆、钼、钌等的干扰,且无法通过线性扣除干扰影响;采用加氧-双四极杆模式测定,上述干扰问题均能得以解决。说明CRC技术存在固有缺陷,即缺乏质量甄别能力。
为克服上述局限,美国Agilent公司于2012年推出三重四极质谱(ICP-MS/MS),其在八极反应池前后均添加四极杆质量分析器,有效过滤进入反应池的离子质量,极大地提高了干扰分辨力[74],在超痕量分析领域具有显著优势。Gil-Díaz等[75]使用103Rh作内标,采用三重四极杆ICP-MS加氧模式,将77Se转化为93SeO进行分析(77Se+16O→93SeO),有效避免了双电荷稀土元素干扰(主要是154Sm++、54Gd++),获得满意结果,回收率为70%~134%(n=3)。Bolea-Fernandez等[76]以氟化甲烷和氦气混合气作反应气,使用ICP-MS/MS分析河口沉积物标准物质(NBS SRM1646)硒含量,分析结果与给定值的绝对误差<0.05。该模式下分析80Se,检出限可低至4ng/L。
(3)同位素质谱分析技术
同位素质谱(IDMS)是分析化学的前沿技术,既可分析元素总量,也可分析形态含量[74],还能用于土壤硒迁移示踪工作[77],是研究土壤硒迁移转化机理的有效手段之一。
硒同位素质谱分析技术可满足高精度分析要求,前提是需进行硒分离纯化,并与具有高分辨力的检测器联用,如多接收器等离子体质谱仪(MC-ICP-MS)[78]。Pons等[79]建立了HG-MC-ICP-MS同位素双稀释剂(76Se-78Se)分析硒的精准方法,硒信号灵敏度超过1000V/ppm,分析误差介于0.01‰~0.025‰。谭德灿等[78]建立了MC-ICP-MS同位素双稀释剂(74Se-77Se)分析硒的精准方法,获得最优长期精准度为0.00‰~0.10‰。由此可见,IDMS可作为应对超低硒分析挑战的有力工具。
2.2.3X射线分析技术
(1) X射线荧光光谱技术
X射线荧光分析法(XRF)因具有绿色无损、分析速度快的特点,在生物、材料分析领域备受青睐。由于土壤硒含量通常较低且基体干扰严重,XRF在该领域分析能力无法与光谱和质谱类分析方法媲美,但与样品预浓缩技术相结合,能提高分析性能。
Marguí等[80]探索了几种分析方法结合全反射XRF测定土壤硒含量的可能性,通过改进样品前处理工艺减少基质效应影响,开发了分散液液微萃取程序,该方法分析土壤硒检出限为0.05mg/kg,与光谱分析技术获得的检出限相当或更低。Kocot等[81]建立了一种分散固相微萃取- XRF测定硒的方法,并将其用于矿物、水体和生物材料硒分析。该方法以石墨烯为固体吸附剂,吡咯烷二硫代氨基甲酸铵(APDC)为螯合剂,采用能量色散X射线荧光光谱法(EDXRF)对无机硒开展了形态分析和测定。在优化条件下,Se(Ⅳ)和Se(Ⅵ)回收率分别为97.7%±5.0%和99.2%±6.6%,精密度(RSD)为5.1%~6.6%。允许获得最低检测限为0.032ng/mL,富集系数高达1013±15。
(2)同步辐射X射线吸收光谱技术
同步辐射X射线吸收光谱技术能够对土壤进行无损原位形态分析,其主要使用X射线吸收近边结构(XANES)区域对固体表面结构进行鉴定,该区域出现在吸收边缘下方-20eV和上方+50eV之间,提供了有关元素氧化状态、相关配体和直接配位环境的信息,能够直接说明土壤硒的具体存在形式。
同步辐射技术能够补充现有形态分析技术的不足。Scheinost等[82]分别使用同步辐射技术和分步提取方法对冶炼厂污染土壤进行形态分析,结果显示相比于同步辐射分析结果,分步提取方法未能识别与若干矿物相相关的物种,显示出同步辐射技术在土壤形态分析方面的优势。Favorito等[50]借助同步辐射X射线吸收光谱技术,评价了分步提取程序对土壤硒形态的分离能力,证实了提取剂提取不完全的现象。
该技术在土壤硒赋存机理研究方面具备强大优势。Qin等[83]综合使用微聚焦X射线荧光光谱技术、X射线衍射技术和X射线吸收精细结构谱技术,分析了废弃尾矿污染土壤中硒的存在形式,得出硒主要以Se(Ⅳ)形式存在,Fe(Ⅲ)氢氧化物是Se(Ⅳ)的宿主相。Nie等[84]通过扩展X射线吸收精细技术分析了Se(Ⅳ)在针铁矿上的吸附行为,发现Se(Ⅳ)在中、低pH环境下形成双齿、强键合内球面配合物,其他物质可通过竞争/协同等作用方式影响其相互作用。
同步辐射技术应用于土壤硒形态分析,需注意X射线本身能够改变样品硒形态。研究发现X射线能够使Se(Ⅵ)迅速还原为Se(Ⅳ)[37]。此外,同步辐射技术对土壤可流动相中物种的鉴定存在不足[40],无法区分吸附到非晶态和晶态铁氧化物上的硒的参考光谱[85]。
3 土壤硒标准物质研制现状
标准物质是用于地球化学样品分析质量控制的重要因素。目前具有硒含量定值的土壤标准物质可覆盖的土壤基体和硒含量范围有限,限制了大规模地球化学勘探和评价中对样品的有效监测。受制于土壤硒形态提取技术的发展,当前针对土壤硒原始形态定值的标准物质和针对分步提取技术定义形态的标准物质的研制工作有待推进。
3.1 国外土壤硒标准物质
表5列出了部分能够通过国家标准物质资源共享平台查询到的国外研制的具备标准物质证书和硒含量定值的土壤标准物质。
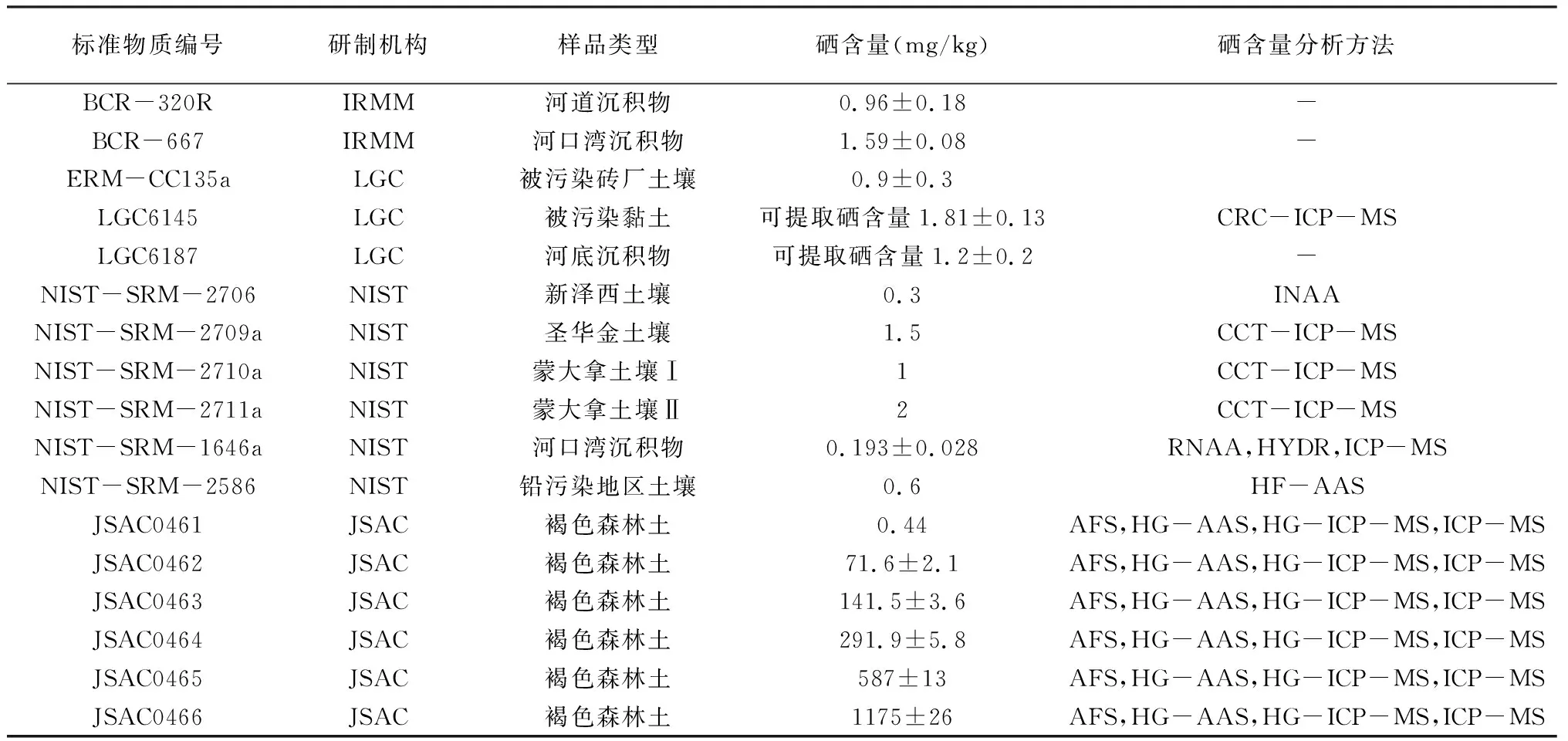
表5 具备硒含量定值的国际土壤标准物质
从表5可以看出,欧洲、美国和日本是国际土壤标准物质的主要来源。相应标准物质全硒含量多在1mg/kg左右。除日本外,其余国家均未使用AFS技术对土壤硒含量进行定值,多采用ICP-MS、AAS和INAA方法。欧盟研制土壤标准物质时较为关注污染类土壤;美国土壤标准物质倾向于按地区进行研制;唯独日本研制的标准物质存在硒含量梯度划分,硒含量范围从0.44mg/kg跨越到1175mg/kg。使用具有相同基体类型、具有梯度含量变化的系列标准物质对仪器进行标化,能够最大限度地降低仪器分析中因样品基体与标准溶液基体差异造成的误差。此类系列标准物质在分析中存在较大需求,也是中国紧缺的类型。
3.2 中国土壤硒标准物质
中国土壤地球化学标准物质研制起步于20世纪70年代末,现已研制了54种土壤成分分析标准物质,相关信息如下。
(1) 35种土壤成分分析标准物质(GBW07401~GBW07408、GBW07423~GBW07430、GBW07446~GBW07457、GBW07385~GBW07391)。均具备全硒含量定值,除GBW07405和GBW07406硒含量大于1mg/kg,其余标准物质硒含量均较低;除GBW07401~GBW07408使用原子荧光光谱法、比色法和极谱法对硒含量进行定值外,其余标准物质均仅使用原子荧光光谱法定值。
(2) 5种土壤形态成分分析国家标准物质,包括:江苏太湖沉积物(GBW07441)、北京土壤(GBW07442)、湖北水稻土(GBW07443)、城市街道尘土(GBW07444)和华南矿化区土壤组合样本(GBW07445),是由国家地质实验测试中心于2008年研制。采用七步顺序提取法分析了土壤水溶态、离子交换态、碳酸盐结合态、腐植酸结合态、铁锰氧化物结合态、强有机结合态、残渣态七种形态,定值了16种元素(Cd、Cu、Pb、Zn、Mo、Co、Cr、Ni、Mn、Fe、Ca、K、As、Hg、Sb、Se)的形态含量和总量,其中硒元素总量最高值为(0.56±0.012)mg/kg,满足了区域地球化学调查及矿产勘查的需要。
(3) 10种具备稀盐酸提取态硒含量定值和水溶态硒含量定值的土壤有效态成分分析标准物质(GBW07412a~GBW07417a,GBW07458~GBW07461):是由中国地质科学院地球物理地球化学勘查研究所于2010年研制。其中,GBW07412a~GBW07417a和GBW07458~GBW07461定值了水溶态硒含量,最高值为(15±5)μg/kg;GBW07412a、GBW07415a~GBW07417a、GBW07458定值了稀盐酸提取态硒含量,最高值为7.7μg/kg。
(4) 4种富硒土壤成分分析标准物质(GBW07900~GBW07904):是由中国地质大学(武汉)于2018年研制,硒含量介于0.83~19.5mg/kg。
4 存在问题和发展展望
人类活动加剧了土壤硒分布异质性,意味着样品类型和含量范围不断变化,要求土壤硒分析技术和标准物质研制能够随之发展,以保证分析质量。
当前土壤硒分析领域存在的问题和发展趋势是:①前处理技术相对仪器分析技术发展较缓,成为限制该领域发展的一大瓶颈。分步提取技术的出现为土壤硒形态提取开辟了新思路,但却未能完全解决提取专一性和提取过程硒形态转化的问题。这仍是一个充满挑战的课题。②质谱技术较光谱技术在痕量、超痕量分析领域具有显著优势,碰撞/反应池技术结合多重四极杆技术理论上能够实现无干扰测定,且检出限低、检测线性范围广,是当前土壤硒分析领域的热点技术。③同步辐射X射线荧光技术具备强大的原位形态分析能力,能在一定程度上弥补前处理技术不足,但其无法鉴定可流动相物种形态,将其作为传统研究方法的补充是土壤硒分析的趋势和要求。④土壤硒标准物质研制滞后于分析技术的发展,无法满足分析质量监控需求,有待加强开展相关研制工作。
致谢:感谢国家标准物质共享平台工作人员在国内外标准物质查询方面给予的指导和帮助。
猜你喜欢
中老年保健(2022年5期)2022-11-25
中老年保健(2022年4期)2022-08-22
中学生数理化·中考版(2022年5期)2022-06-05
中学生数理化·中考版(2021年5期)2021-11-22
食品安全导刊(2021年20期)2021-08-30
红领巾·探索(2021年2期)2021-08-26
学生天地(2020年34期)2020-06-09
当代陕西(2019年11期)2019-06-24
作文通讯·高中版(2017年12期)2017-02-06
当代化工研究(2016年5期)2016-03-20
