
钒基催化剂同步去除燃煤烟气中甲苯与NOx研究
2021-09-03 07:13张益兰肖高飞李剑晗杜玥莹付名利华南理工大学环境与能源学院广东广州50006广东省大气环境与污染控制重点实验室广东广州50006广东省环境风险防范与应急处置工程技术研究中心广东广州50006
中国环境科学 2021年8期
张益兰,肖高飞,李剑晗,杜玥莹,付名利,2,3,胡 芸,2,3* (.华南理工大学环境与能源学院,广东 广州 50006;2.广东省大气环境与污染控制重点实验室,广东 广州 50006;3.广东省环境风险防范与应急处置工程技术研究中心,广东 广州50006)
燃煤过程产生的有机污染物以挥发性有机物为主[1-2],是臭氧和PM2.5的重要前驱体[3-6],对生态环境构成重大威胁.国内外有关燃煤过程中有机污染物生成机理认识不足,更缺乏相应控制手段.相关研究表明燃煤电厂及火电厂排放的主要VOCs成分为苯、甲苯、乙苯及邻二甲苯[7-8].与工业源 VOCs相比,燃煤烟气中的VOCs具有成分复杂、浓度低、烟气量大、毒性高等特点[9-11],无法应用现有工业源VOCs催化氧化技术.基于传统 SCR配方设计多污染物控制催化剂是更有应用前景的方向.鉴于此,本文拟将脱硝技术与有机物氧化技术联用,期望研发针对燃煤电厂脱硝区内高效的抗水热老化、抗中毒和耐机械磨损的催化剂,同时脱除有机物和NO.
催化燃烧的操作温度为200~450℃,采用该方法对宽浓度范围内的有机废气进行处理是目前应用最广泛的有机物控制技术[12-17],且与 SCR的工艺段温度基本一致,其关键是合适的催化剂材料.有研究表明,目前商业上常用的 V2O5-WO3/TiO2催化剂对VOCs的去除有一定效果[18-19].然而,V2O5-WO3/TiO2催化剂在反应过程中生成了大量有毒副产物,COx选择性很低[20-21].为了改善其催化氧化性能,其他活性金属组分如 Mo、Ce、Mn、Co、Zr等常被用来修改典型的 SCR催化剂配方.有研究表明,Mo-Ce/TiO2催化剂在模拟SCR气氛下具有出色的同时还原NO和氧化Hg(0)的效率[22].掺杂适量的Mo可以帮助增加比表面积,形成更多可还原的物种,有效提高Mn/TiO2的SCR催化性能,同时具有更好的 SO2耐受性[23].因此,本文尝试采用 Mo改性钒基催化剂,以提高V2O5-WO3/TiO2的氧化效率.
目前商业SCR催化剂主要有蜂窝式、平板式和波纹板式三种形状[24].堇青石蜂窝陶瓷机械强度高、热膨胀系数小,已经被广泛作为蜂窝状涂覆型催化剂的基体材料,但其涂层负载量有限且易脱落[25-26],需要选择合适的粘结剂和制备工艺.本文采用不同的工艺制备了V2O5-WO3-MoOx/TiO2堇青石整体式催化剂,以甲苯和NO为探针分子,考察了Mo的添加量、涂覆方法、粘结剂的种类等制备工艺对整体式催化剂活性的影响,并用XRD、SEM-EDS、FT-IR、BET等对催化剂进行了表征.本文可作为复杂燃煤烟气中VOCs和NOx的协同治理技术提供了新材料,同时为整体式催化剂在燃煤电厂中的实际应用提供了理论基础和实验积累.
1 材料与方法
1.1 催化剂的制备
1.1.1 V2O5-WO3-MoOx/TiO2-粉末催化剂 将0.8000g草酸、0.0459g偏钒酸铵、0.1656g钨酸铵、0.1104g钼酸铵加入20mL去离子水中,在水浴锅中60℃恒温搅拌,加入2g锐钛矿TiO2,继续60℃恒温搅拌至蒸干,干燥(105℃,3h),煅烧(500℃,5h),得到V2O5-WO3-MoOx/TiO2粉末.根据需要添加不同比例的 Mo元素(V1W6Mo1/TiO2、V1W6Mo3/TiO2、V1W6Mo6/TiO2、V1W6Mo9/TiO2),标记为VxWyMoz/TiO2.整体式催化剂浸渍液配比采用粉末状催化剂的最佳比例.
1.1.2 V2O5-WO3-MoOx/TiO2-堇青石整体式催化剂 堇青石预处理:将堇青石切割成直径为 25mm,高为8mm的样品块.将其置于质量分数为28%的硝酸中浸泡4h后洗涤、吹脱残液、干燥(120℃,3h)、煅烧(500℃,5h),去除表面杂质.
一次浸渍法:将2.0000g草酸,0.4560g偏钒酸铵、1.4467g钨酸铵、1.1040g钼酸铵加入到60mL去离子水中,60℃恒温搅拌,加入20g锐钛矿TiO2,形成稳定的 V2O5-WO3-MoOx浆液.将经预处理的堇青石浸渍于浆液中超声负载,经干燥(120℃,3h)和煅烧(500℃,5h),得到的催化剂标记为 VWMo/TiO2-堇青石-一次浸渍.
涂敷法:在上述 V2O5-WO3-MoOx浆液制备过程中加入质量分数为1%的粘结剂(硅溶胶、拟薄水铝石或甲基纤维素),其余步骤相同.添加硅溶胶、拟薄水铝石、甲基纤维素的堇青石整体式催化剂分别标记为VWMo/TiO2-堇青石-SSO、VWMo/TiO2-堇青石-PB、VWMo/TiO2-堇青石-MC.
分步浸渍法:参见一次浸渍法步骤制得 V2O5-WO3-MoOx浆液和TiO2浆液.将样品置于TiO2浆液中超声负载 10min,经负载干燥煅烧后将 TiO2涂层负载在载体上,采用同样的步骤再负载 V2O5-WO3-MoOx涂层,得到催化剂VWMo/ TiO2-堇青石-分步浸渍.
溶胶凝胶法:将样品置于纳米 TiO2溶胶中超声负载10min,经负载干燥煅烧将TiO2负载在载体上,参见分步浸渍法,继续负载V2O5-WO3-MoOx涂层,得到催化剂VWMo/TiO2-堇青石-溶胶凝胶.
1.2 催化剂性能测试
催化剂活性测试在固定床反应器中进行(图1),粉末催化剂反应器为一根内径6mm、长度45cm的石英管,催化剂经造粒处理(40~60目),每次装入催化剂 0.1g,反应的质量空速 120000mL/(g·h);整体式蜂窝陶瓷催化剂的反应器为一根外径30mm、内径28mm、长度为70cm的石英管.体积空速为3000h-1,反应温度(150~390℃)由K型热电偶控制.测试条件为:[SO2]=1000×10-6, [NO]=500×10-6, [NH3]=500×10-6, [O2]=5vol%, N2为平衡气体, H2O的体积分数为 5v%.检测系统为一台配有 2个氢火焰离子化检测器(FID)和甲烷转化炉的气相色谱仪(检测甲苯、CO及CO2).加水和模拟烟气氛围的测试由MKS公司的 MultiGas2030红外傅立叶变换光谱分析仪完成,在线同时检测甲苯、NO、NH3、SO2、H2O的实时浓度值,各气体组分每 5s取一个数值.测试流程:先在旁路稳定好温度,接着通入甲苯、NO、NH3、SO2,各组分浓度稳定并达到反应温度后再加入5v%的H2O.

图1 评价测试装置示意Fig.1 Schematic of experimental set-up
甲苯转化率(Xtoluene)和 COx(CO、CO2)选择性( S C Ox)分别可通过公式(1)、(2)得到:

1.3 催化剂的表征.
通过X射线衍射(XRD)来测定各催化材料的物相结构,所采用的仪器为D8ADVANCE型X射线衍射仪(Bruker-AXS公司,德国),使用 ASAP 2020M 全自动表面分析仪(Micromeritics,美国)测试样品比表面积,采用静态吸附法,在液氮恒温条件下(77K)进行测试,得到吸附脱附等温线及 BJH吸脱附孔体积分布曲线,样品的比表面积使用Brunauer-Emmett-Teller方程计算获得.扫描电子显微镜(SEM)为 Ultra55型仪器,仪器测试电压为15kV,电流为10mA.FT-IR图谱由Nicolet-6700型傅立叶变换红外光谱仪测得,扫描范围 4000~400cm-1,分辨率为1.93cm-1,扫描次数64次.
2 结果与讨论
2.1 催化剂的制备方法优化与性能评价
2.1.1 催化剂配方的确定 将不同 Mo负载量的VWMo/TiO2与VW/TiO2、VMo/TiO2催化氧化甲苯性能进行比较,由图 2可知,V1W6Mo3/TiO2具有最佳活性和选择性,其达到 90%甲苯去除率时的温度(T90)为 287℃,在 350℃的选择性(S350)为 98%.相较于VW/TiO2(T90=327℃)和 VMo/TiO2(T90=324℃)以及市售商业催化剂(T90>390℃),添加 Mo之后的VWMo/TiO2(T90=287~308℃)对甲苯的催化氧化性能有了明显的提升.在另一项研究中[27],Ce/Mo掺杂改性的V-W/Ti型催化剂在SCR工作温度窗口有着良好的苯和甲苯去除效果,实现去除NO和VOCs的协同作用(T=260~420℃).
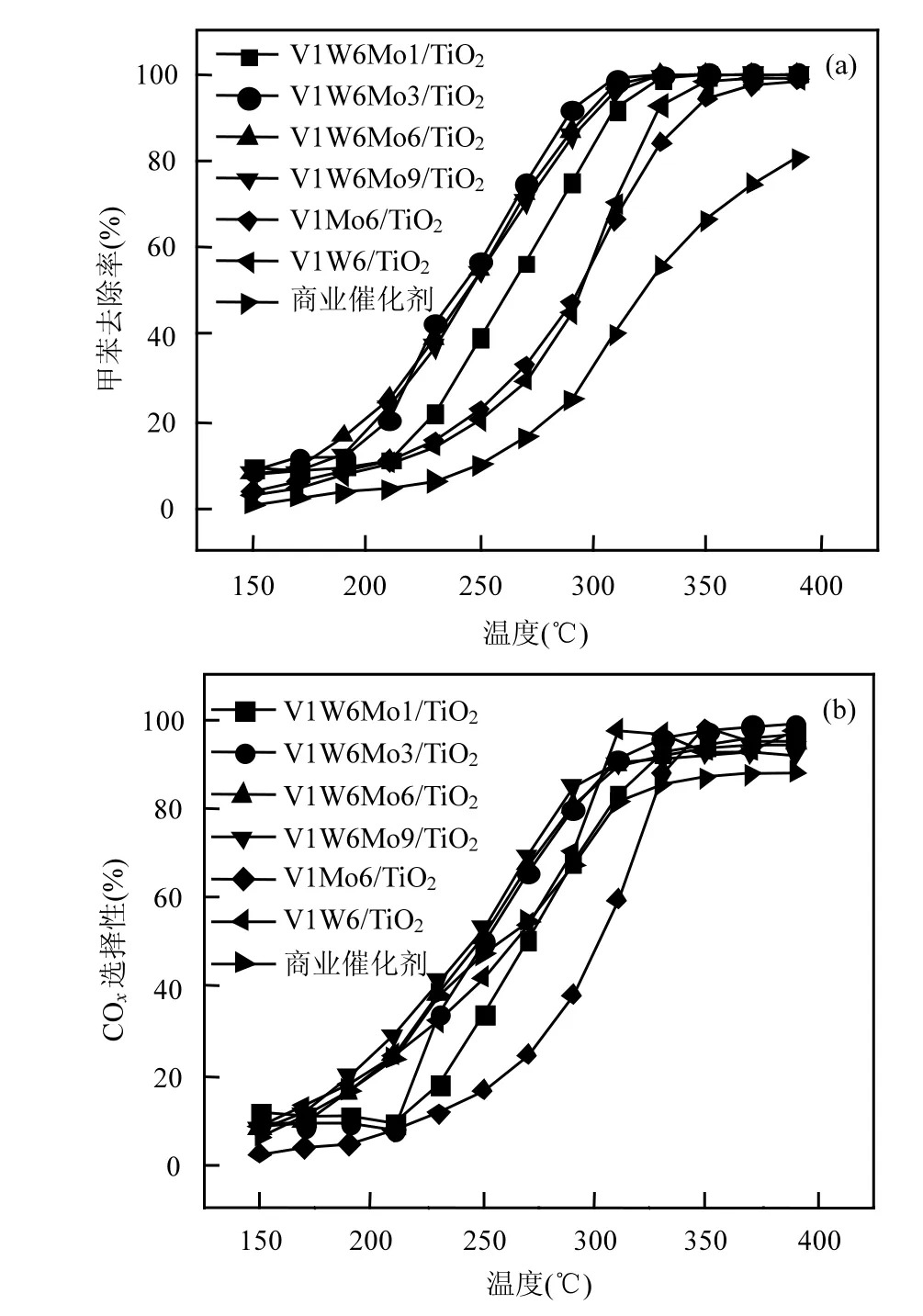
图2 不同Mo负载量的催化剂催化氧化甲苯性能比较Fig.2 Comparison of toluene oxidation performance over catalysts with different Mo loads
2.1.2 O2对催化去除甲苯和 NO性能的影响 如图3(a),当没有O2时,催化剂几乎不降解甲苯.O2含量为 1%时,催化剂活性迅速提高到 94%.当 O2含量达到 3%后,随着 O2浓度的增加,催化剂去除甲苯的效率不再有明显变化.如图 3(b)所示,当 O2含量为0%时,催化剂的活性较低,去除NO的效率仅37%;当 O2含量为 1%时,催化剂活性提高到 97%,O2含量继续增加时,催化剂去除甲苯的性能也不再有显著变化.

图3 O2对V1W6Mo3/TiO2去除甲苯和NO性能影响Fig.3 Effect of O2 on the removal of toluene and NO by V1W6Mo3/TiO2
2.1.3 甲苯与NO在燃煤烟气中的同步去除性能燃煤烟气中环境成分复杂,含有高浓度的 NH3、NO、SO2等污染物,因此考察了催化剂在模拟燃煤烟气(NH3、NO、SO2)下对甲苯与 NO同步去除性能.由图4(a)和4(b)可看出,未改性的V1W6/TiO2催化剂的甲苯转化率很快下降,而 V1W6Mo3/TiO2催化剂在350℃时有优异的甲苯去除率和 NO去除率,24h反应后对甲苯去除率、NO去除率均为99%.
水在燃煤烟气中也是一种重要组分,因此考察了催化剂对甲苯和NO的去除率及N2选择性(NO、NH3、SO2、及 H2O 同时存在).如图 4(c)和 4(d),H2O和其他烟气组分同时加入时,甲苯去除率、NO去除率和 N2选择性未降低,反而有一定的提高.表明V1W6Mo3/TiO2催化剂在烟气环境中可同步脱除NO和甲苯,并且具有优异的抗烟气稳定性.有研究表明[28],适量的Mo作为助催化剂,可以提高催化剂对H2S的竞争吸附能力,从而减缓活性中心的硫化速率,有助于提高催化剂的抗硫性能,这可能是本文 Mo改性催化剂具有优异的抗烟气稳定性的部分原因.
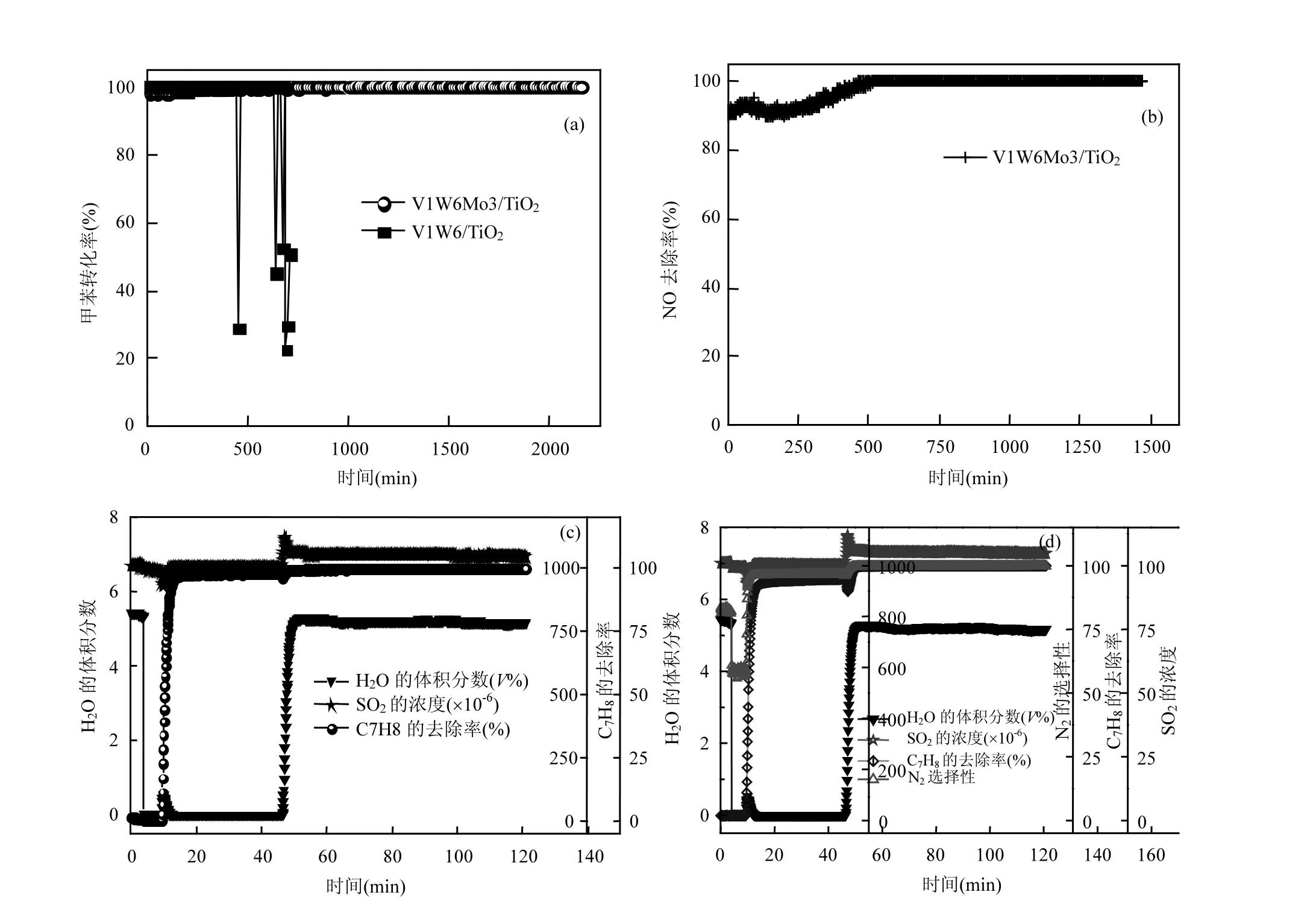
图4 V1W6Mo3/TiO2催化剂催化氧化甲苯稳定性Fig.4 Stability of V1W6Mo3/TiO2 catalyst for toluene oxidation
2.1.4 制备方法对整体式催化剂催化氧化甲苯性能的影响 如图 5所示,VWMo/TiO2-堇青石-一步浸渍法和 VWMo/TiO2-堇青石-涂敷法均表现出较高的甲苯转化率和COx选择性,T90分别为298和321℃.这表明随着粘结剂的加入,催化剂活性有所降低.考虑到除活性外,机械稳定性也是其重要指标,对催化剂的机械稳定性进行了考察.VWMo/TiO2-堇青石-一步浸渍法和VWMo/TiO2-堇青石-涂敷法制备的催化剂的负载量分别为16.11和28.26%.超声处理后 VWMo/TiO2-堇青石-一步浸渍法的质量损失较多,脱落率为 24.93%,而 VWMo/TiO2-堇青石-涂敷法的质量损失仅为 6.81%.结合催化剂活性和机械稳定性,综合考虑,选取涂敷法来制备 VWMo/TiO2-堇青石整体式催化剂.

图5 制备方法对整体式催化剂催化氧化性能的影响Fig.5 Effect of preparation method on catalytic oxidation performance of monolithic catalyst
2.1.5 不同粘结剂对整体式催化剂抗 SO2性能的影响 图 6(a),6(b)显示了不同粘结剂(硅溶胶(SSO)、拟薄水铝石(PB)、甲基纤维素(MC))在添加量为 1%时所制得的催化剂催化氧化甲苯的性能和COx选择性,其中 VWMo/TiO2-堇青石-MC 的性能最佳(T90=307℃),其次为 VWMo/TiO2-堇青石-SSO(T90=320℃), VWMo/TiO2-堇青石-PB的性能最差(T90=326℃).研究表明在烟气环境中,SO2的存在可能对催化剂的催化氧化性能产生显著的抑制作用[29-30],因此考察了在 1000×10-6SO2存在的情况下,不同粘结剂的整体式催化剂去除甲苯的效率(图6(c)、6(d)).如表1所示,VWMo/TiO2-堇青石-MC表现出优异的抗硫性能,添加 1000×10-6SO2前后,催化剂的 T90几乎不变, VWMo/TiO2-堇青石-SSO、VWMo/TiO2-堇青石-PB的 T90明显升高,表明 MC为最佳粘结剂.

表1 不同粘结剂类型的整体式催化剂T90对比Table 1 Comparison of monolithic catalyst T90 with different binder types
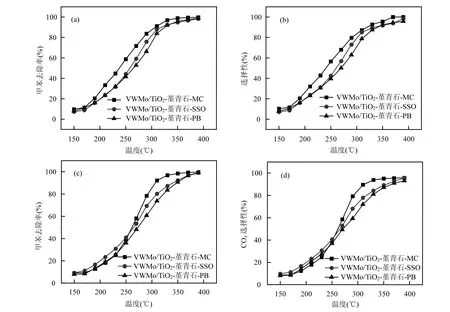
图6 不同粘结剂类型的整体式催化剂催化氧化甲苯性能对比Fig.6 Comparison of toluene oxidation performance for monolithic catalysts with different kinds of binder
2.2 催化剂的表征
2.2.1 催化剂的结构表征 如图 7所示,在 2θ=25.4°、37.8°、48.0°、54.0°、55.1°和 62.8 °处都出现了典型的 TiO2锐钛矿结构的(101)、(004)、(200)、(105)、(211)和(204)晶面特征衍射峰[31],其特征衍射峰与标准卡片一致(JCPDSNO.21-1272),说明VWMo/TiO2材料涂覆在堇青石上并未影响载体的晶面结构.XRD谱图中并未发现V、Mo、W的特征衍射峰,表明活性组分V、Mo、W的含量少且高度分散.样品比表面积及孔容孔径的大小规律与活性规律并不吻合,表明比表面积及孔容孔径不是影响催化剂性能的主要因素(表2).
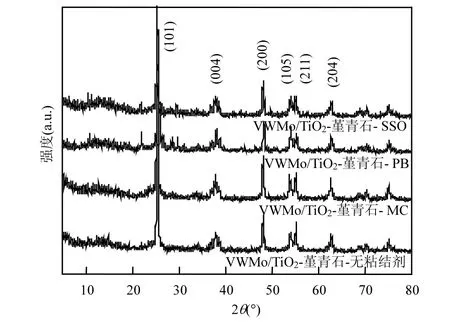
图7 不同粘结剂整体式催化剂XRD谱图Fig.7 XRD patterns of the monolithic catalysts with different binders

表2 样品比表面积及孔容孔径Table 2 Specific surface area, pore volume and pore diameter of the samples
图8为采用不同粘结剂制备的整体式催化剂的吸附脱附等温线,不添加粘结剂时比表面积和孔体积最大,添加粘结剂会堵塞一定的孔道结构,降低其比表面积和孔体积.

图8 不同粘结剂整体式催化剂吸附脱附等温线Fig.8 Adsorption-desorption isotherm of monolithic catalysts with different binders
不同粘结剂制备的整体式催化剂放大50000倍的SEM图像如图9(A)所示,可以看出催化剂呈现颗粒团簇状,主要是由形状规则的微粒组成.图 9(B)为催化剂的 EDS扫描,表明采用不同粘结剂的整体式催化剂均含有Ti、O、V、Mo、W5种元素.通过图9(C)的mapping扫描表明Ti、O、V、Mo、W元素在催化剂表面高度分散.


图9 不同粘结剂整体式催化剂SEM-EDS图Fig.9 SEM-EDS-mapping of monolithic catalysts with different binders
由表 3可知,经过超声处理后,不添加粘结剂的整体式催化剂质量损失最多,为 25.74%.超声震荡90min后,VWMo/TiO2-堇青石-MC的质量损失仅为2.45%,其耐磨性能在3种粘结剂中最好.
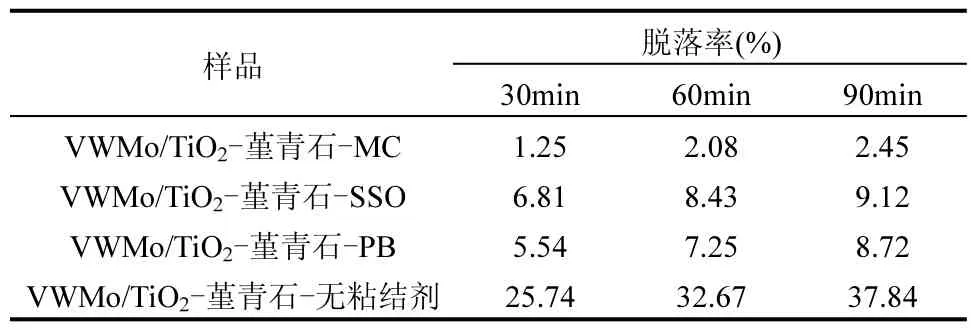
表3 不同粘结剂类型的整体式催化剂脱落率随超声震荡时间变化Table 3 The abscission rate of monolithic catalyst with different binder types varies with the ultrasonic oscillation time
2.2.2 催化剂的红外光谱分析 如图10所示,位于771cm-1处的吸附峰可以归属于锐钛矿TiO2[32],位于957和 1095cm-1处的吸附峰分别归属于 V-O[33]和V=O[34]的伸缩振动,位于 1606cm-1处的峰归属于催化剂表面吸附水的伸缩振动[35].上述吸附峰的出现表明VWMo/TiO2-堇青石催化剂顺利合成.
从图10(a)可以看出, 添加粘结剂并不会影响催化剂的本征结构.比较反应前后红外光谱图的变化,本文制备的VWMo/TiO2-堇青石催化剂在反应前后活性组分的吸附峰略有增强,在以甲基纤维素和硅溶胶作为粘结剂的催化剂中观察到类似的现象,表明上述材料具有优异的抗硫稳定性.其中MC作为粘结剂的催化剂与无粘结剂时的红外谱图具有高度一致性,从图 10(b)也可以看出,即使是在 1000×10-6SO2的氛围中反应 24h后,该催化剂的红外谱图仍保持稳定,可见甲基纤维素不会影响催化剂的活性.然而,当用拟薄水铝石作为粘结剂时,反应前后催化剂表面活性组分的吸附峰强度明显下降,这表明在耐硫活性评价之后该催化剂出现失活现象.原因是拟薄水铝石含有氧化铝,催化剂上的积硫会氧化为SO3,SO3会与氧化铝反应生成硫酸铝而使催化剂失活[36],而甲基纤维素不含氧化铝,所以它作为粘结剂所制备的催化剂活性更好.以上分析结果与活性评价的结果相一致,即 VWMo/TiO2-堇青石-无粘结剂≈VWMo/TiO2-堇青石-MC>VWMo/TiO2-堇青石-SSO>VWMo/TiO2-堇青石-PB.
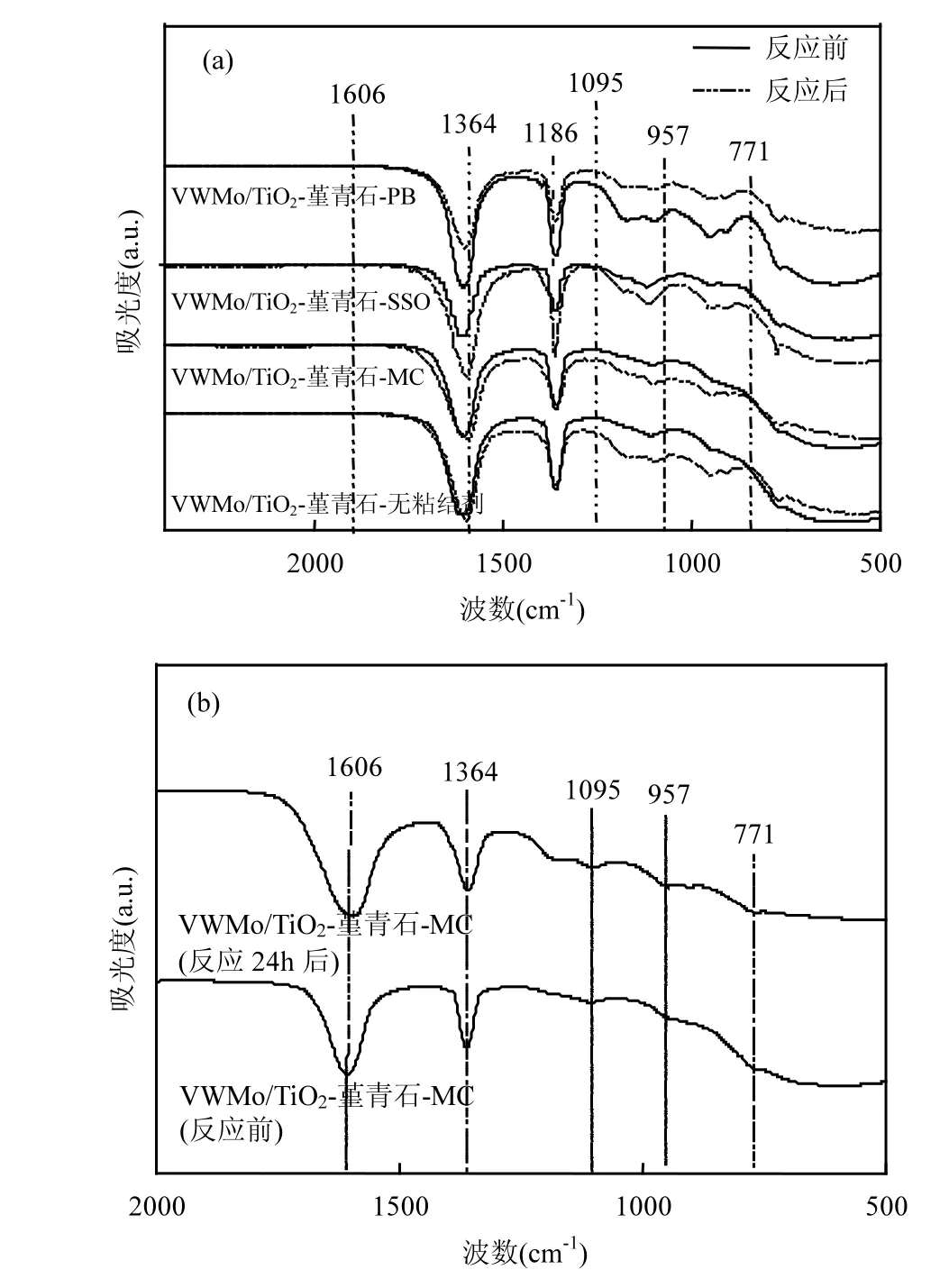
图10 堇青石催化剂反应前后的红外谱Fig.10 FT-IR spectra of cordierite catalysts before and after reaction
3 结论
3.1 V1W6Mo3/TiO2粉末催化剂在烟气环境中可同步脱除 NO和甲苯,并且具有优异的抗烟气稳定性.当反应温度为 350℃,H2O、NO、NH3和 SO2等烟气组分同时存在时,催化剂催化氧化甲苯去除率可达99%,NO去除率为100%,N2选择性为99%.
3.2 采用不同制备方法将其制成堇青石蜂窝陶瓷整体式催化剂,结合催化剂活性和机械稳定性,涂敷法制备的 VWMo/TiO2-堇青石-MC整体式催化剂具有最优活性(T90=307℃)和机械稳定性(负载率=28.26%,脱落率=6.81%).
3.3 XRD、SEM-EDS表明V、Mo、W活性组分在分布均匀且高度分散.FT-IR显示VWMo/TiO2-堇青石-MC整体式催化剂在 1000×10-6SO2的氛围中反应 24h后,其红外谱图仍然保持稳定,表明其具有优异的抗硫性能.
猜你喜欢
现代装饰(2022年5期)2022-10-13
雪豆月读·高年级(2022年4期)2022-09-15
黑龙江交通科技(2021年10期)2021-11-01
陶瓷学报(2021年4期)2021-10-14
黑龙江交通科技(2021年9期)2021-10-13
中华养生保健(2020年9期)2021-01-18
石油沥青(2019年3期)2019-07-16
无机化学学报(2019年2期)2019-02-27
宝藏(2018年1期)2018-01-31
腐植酸(2016年1期)2016-12-16
