
药用四水碳酸镧及其杂质碱式碳酸镧的定性和定量分析
2021-09-01 12:37:14马雨璇
分析仪器 2021年4期
马雨璇 王 娜 李 钢
(南京师范大学食品与制药工程学院, 分析测试中心, 南京 210023)
慢性肾脏病(CKD)的发病率与日俱增,患者普遍伴随着钙磷代谢紊乱问题,肾脏排磷量显著减少会导致高磷血症。高磷血症是维持性血液透析患者死亡的独立危险因素,已经成为慢性肾疾病的常见并发症[1,2]。传统的铝盐和钙盐磷结合剂由于其毒副作用大,已经被弃用。镧是稀土中的阳离子元素,以三价形式(La3+)存在,对羧基和磷酸基具有较强的亲和力,可与磷酸盐结合形成水溶性低的磷酸镧难溶物。目前被开发成药物碳酸镧(LC),于2004年10月由美国FDA批准应用于临床[3-5]。碳酸镧主要是通过对细胞内的氧化应激反应进行抑制,激活细胞外信号调节激酶来达到抑制内皮细胞凋亡的目的[6]。该复合镧盐在胃肠道吸收少,主要经胆汁排泄,少部分经肠道排泄,肾脏排泄甚少,所以对肾病患者不造成肾脏的二次损伤[7,8]。经过长期临床试验表明,虽然碳酸镧与碳酸钙在磷结合率上效用相当,但是长期使用钙类磷结合剂可导致高钙血症,增加血液中的钙磷乘积以及心血管疾病的患病风险[9-12],而碳酸镧不仅可降低血液中的钙磷乘积,还可以减少高钙血症和甲状旁腺激素相关疾病的发生。临床数据显示碳酸镧耐受性好且副作用少,患者在服用药物后,血磷水平得到明显降低,仅少部分出现一些恶心、呕吐以及便秘的症状[13]。与其他磷结合剂相比,碳酸镧毒副作用小,结合率高,不经肾排泄,具有广阔的市场和良好的前景[8]。
基于现有的相关文献报道并不多,在国内对该药物的研究也比较少,需要对其进行深入的研究。碳酸镧的制作工艺较为复杂,镧离子为硬路易斯酸,易与氢氧根离子结合,制备时需要控制pH值在酸性条件下,将氯化镧(LaCl3)溶液倒入碳酸氢钠(NaHCO3)溶液中形成晶核,再以晶核为中心逐渐扩大形成碳酸镧结晶,其中以4个结晶水的碳酸镧(La2(CO3)3·4H2O)结晶生物利用度最高,药效最好[14]。若制备过程中反应条件控制不当会导致目标碳酸镧药物中混有杂质晶型碱式碳酸镧(LaCO3OH),而美国FDA从未批准其用于高磷血症的临床治疗[14]。为了确保药品质量,需要一种切实有效的方法对La2(CO3)3·4H2O和 LaCO3OH进行定性分析,其中LaCO3OH有I型和II型两种晶型。对于药物不同晶型的研究通常借助于X-射线粉末衍射法(PXRD)、热分析法、傅里叶变换红外光谱法(FT-IR)、扫描电子显微镜法(SEM),在相关文献中已有较多报道[15-17]。本研究主要通过上述现代分析手段对La2(CO3)3·4H2O和LaCO3OH的两种杂质晶型进行表征,建立了定性和定量分析方法,为我国将来仿制药的生产和质量控制提供保障和科学依据。
1 仪器与材料
FA2004型电子天平;SHB-3型循环水式多用真空泵;D/max 2500VL/PC型阳极转靶X射线衍射仪;JSM-5610型扫描电子显微镜;Analysis Diamond热重分析仪;DHG-9070A(101A-1S)鼓风干燥器;VERTEX70 型傅里叶变换红外光谱仪。
La2(CO3)3·4H2O和LaCO3OH晶型I、II由南京某公司提供。
2 碳酸镧样品的定性和定量分析
2.1 定性分析
2.1.1热重-差热分析(TG-DTA)
称取样品约2.0mg 放入铂金坩埚,将坩埚放入 TGA 中进行测试,以氮气为保护气,升温速率为 10 k·min-1,升温范围为 25℃~800 ℃,进行热重分析。
2.1.2傅里叶变换红外光谱法分析(FTIR)
采用KBr压片法检测样品,扫描范围4000 ~ 400 cm-1,分辨率4 cm-1,扫描次数32次,用聚苯乙烯膜校正。
2.1.3扫描电子显微镜法分析(SEM)
取样品粘结在样品座上,镀膜处理后放置于扫描电子显微镜下观察形态结构。测定条件:加速电压8kV,放大倍率2000,样品台直径30mm。
2.1.4X-射线粉末衍射法分析(PXRD)
测定条件为管压:40kV, 管流:100 mA,Cu-Kα辐射,石墨弯晶单色器,发射狭缝(DS)=防散射狭缝(SS)=1°,接收狭缝(RS) = 0.15 mm。扫描速度:5°·min-1,连续扫描,扫描范围3°~ 40°,步宽:0.02°。
2.2 定量分析
2.2.1实验方法
单峰法[17]。
2.2.2X-射线粉末衍射法分析(PXRD)
测定条件为管压:40kV;管流:150 mA,Cu-Kα辐射,石墨弯晶单色器,发射狭缝(DS)=防散射狭缝(SS)=1°,接收狭缝(RS) = 0.15 mm;扫描速度:1°·min-1;步进扫描,扫描范围10°~ 30°;步宽:0.02°。
2.2.3检测限和定量限
利用检测限(LOD)和定量限(LOQ)两个指标来测试峰面积法和峰高法的灵敏度[18]。计算公式见式(1)和式(2):
LOD=3.3σ/S
(1)
LOQ=10σ/S
(2)
式中σ为标准误差,使用的是标准曲线截距的标准误差;S为标准曲线的斜率。实验中使用origin软件对标准曲线(峰面积法和峰高法)进行计算和分析。
3 结果分析
3.1 定性分析
3.1.1热重-差热分析
图1为四水碳酸镧、碱式碳酸镧晶型I和II样品的热重分析图谱(A: La2(CO3)3·4H2O、B: LaCO3OH-I、C: LaCO3OH-II)。四水碳酸镧在高温下分解主要分为3个步骤,如式(3):
La2(CO3)3·4H2O → La2(CO3)3→ La2O2CO3→ La2O3
(3)
如图1A所示,四水碳酸镧样品在25~350℃发生第一步分解,失去4个结晶水分子,失重比例为17.0%,放出热量141.8J;在350~600℃发生第二步分解反应,失去2个CO2分子,失重比例为17.1%,放出热量281.9J;在600~800℃发生第三步分解反应,失去最后1个CO2分子,失重比例为7.9%,放出热量540.3J,最后的剩余样品为La2O3。其三步的热重损失比例测量值与理论计算值基本一致,说明该样品确为四水碳酸镧,符合其高温分解式。
碱式碳酸镧晶型I和II的高温分解式分为两步,如式(4)和(5):
2LaCO3OH → La2O2CO3+ H2O + CO2
(4)
La2O2CO3→ La2O3+ CO2
(5)
如图1B所示,碱式碳酸镧晶型I在310~590℃阶段发生第一步分解反应,失去1个H2O分子和1个CO2分子,失重比例为16.1%,放出热量382.1J;在590~800℃发生第二步分解反应,失去1个CO2分子,失重比例为9.3%,放出热量1190.0J。该失重测量值与理论计算值基本一致,符合式(4)和式(5)的分解式。
如图1C所示,碱式碳酸镧晶型II在340~580℃阶段发生第一步分解反应,同样也是失去1个H2O分子和1个CO2分子,失重比例为17.6%,放出热量561.8J;第二步分解反应发生在580~800℃时,失去 1个CO2分子,失重比例为9.8%,放出热量722.7J。碱式碳酸镧晶型II的热重损失量符合理论计算值和分解式。
3.1.2傅里叶变换红外光谱分析
图2为四水碳酸镧、碱式碳酸镧晶型I和II样品的FTIR图(A:La2(CO3)3·4H2O、B: LaCO3OH-I、C: LaCO3OH-II),表1为碱式碳酸镧中各官能团的红外特征吸收。由于碱式碳酸镧中还含有-OH,除了归属CO32-的特征峰以外还应该含有可归属于OH的特征峰位置。
图2A中各个吸收峰:3259cm-1处的宽峰为四水碳酸镧中结晶水的O-H伸缩振动和H-O-H弯曲振动所引起的;1470 cm-1和1365 cm-1处归属于CO32-基团的伸缩振动;849cm-1、747cm-1和677cm-1处归属于CO32-基团的变形振动。
根据文献[19]可知,碱式碳酸镧晶体中CO32-的四个吸收峰主要是在1070~1100 cm-1(ν1)、850~880 cm-1(ν2)、1400~1510 cm-1(ν3)和690~730 cm-1(ν4)处。
图2B中各个吸收峰:1499 cm-1和1435 cm-1处的尖峰可归属于ν3,1080 cm-1可归属于ν1,848 cm-1和872cm-1两个吸收峰归属于ν2,而70 cm-1处的特征峰归属于ν4,它们均是由样品中的CO32-基团的变形振动所引起的;3616 cm-1、3485 cm-1处的尖峰归属于自由OH的伸缩振动,590 cm-1处的尖峰归属于OH的弯曲振动。以上的吸收峰均是由碱式碳酸镧晶型I中的各个官能团所引发的特征吸收,可作为其鉴别的特征峰。
如图2C所示,1488 cm-1和1421 cm-1处的尖峰可归属于ν3,1074 cm-1可归属于ν1,858 cm-1和808 cm-1两个吸收峰归属于ν2,而725 cm-1和697 cm-1处的特征峰归属于ν4,它们也是由样品中的CO32-基团的变形振动所引起的;3450 cm-1处的尖峰由自由OH的伸缩振动引起,未看见低波数区的弯曲振动。这些吸收峰均是由于碱式碳酸镧晶型II中的各个官能团所引发的特征吸收,同样也可用作其鉴别的特征峰。
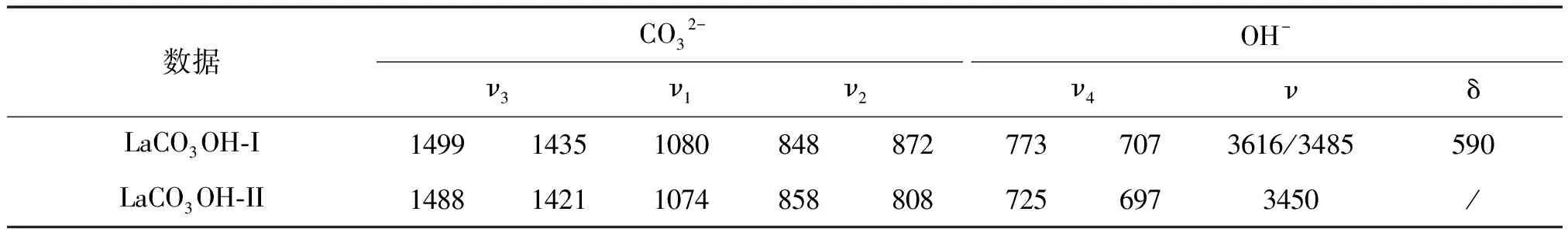
表1 碱式碳酸镧晶型I和II的红外数据

图2 四水碳酸镧、碱式碳酸镧晶型I和II的红外分析图A. 四水碳酸镧; B.碱式碳酸镧晶型I ; C.碱式碳酸镧晶型II
碱式碳酸镧晶型II相比于晶型I,其官能团所引起的吸收特征峰没那么复杂,少了两个自由OH的吸收峰(3616cm-1处OH的伸缩振动和590 cm-1处OH的弯曲振动)。可能是由于晶型结构不同,晶型II中CO32-吸收的贡献占据主要部分,其峰强度更高。从图表中也可以看出碱式碳酸镧晶型II中的CO32-的特征吸收更强、特征峰更清晰尖锐。
3.1.3扫描电子显微分析
四水碳酸镧、碱式碳酸镧晶型I和II样品的扫描电镜微形貌如图3所示(A:La2(CO3)3·4H2O、B:LaCO3OH-I、C:LaCO3OH-II)。图3A可以看出四水碳酸镧的微观形貌为片层状;图3B显示碱式碳酸镧晶型I的微观形貌是大小均匀的颗粒状;图3C是碱式碳酸镧II其微观形貌为团聚在一起大小不一的絮状。

图3 四水碳酸镧、碱式碳酸镧晶型I和II的扫描电镜图A. 四水碳酸镧; B. 碱式碳酸镧晶型I ; C. 碱式碳酸镧晶型II
3.1.4X-射线粉末衍射分析
从图4中可以看出,LaCO3OH的I型和II型与La2(CO3)3·4H2O各自有不同的特征峰。La2(CO3)3·4H2O(A)在2θ=13.4°、18.32°、19.66°、22.92°、23.92°、27.04°和30.34°处有特征峰,2θ=13.4°处为最强峰;LaCO3OH的I型(B)在2θ=17.62°、24.3°、30.22°和35.78°处有特征峰,2θ=30.22°处为最强峰;LaCO3OH的II型(C)在2θ=15.68°、20.26°、20.64°、23.58°、23.9°、26.1°、29.8°、31.84°、33.5°、35.37°、36.54°和37.82°处有特征峰,2θ=29.8°处为最强峰。结果与PDF卡片基本一致,误差在±0.2°范围内,可据此来鉴别3种晶型。同时,不同的特征峰也可作为LaCO3OH的I型和II型与La2(CO3)3·4H2O定量分析的基础,表2~表4中加粗部分代表后续定量分析3种晶型所选取的特征峰结构数据。
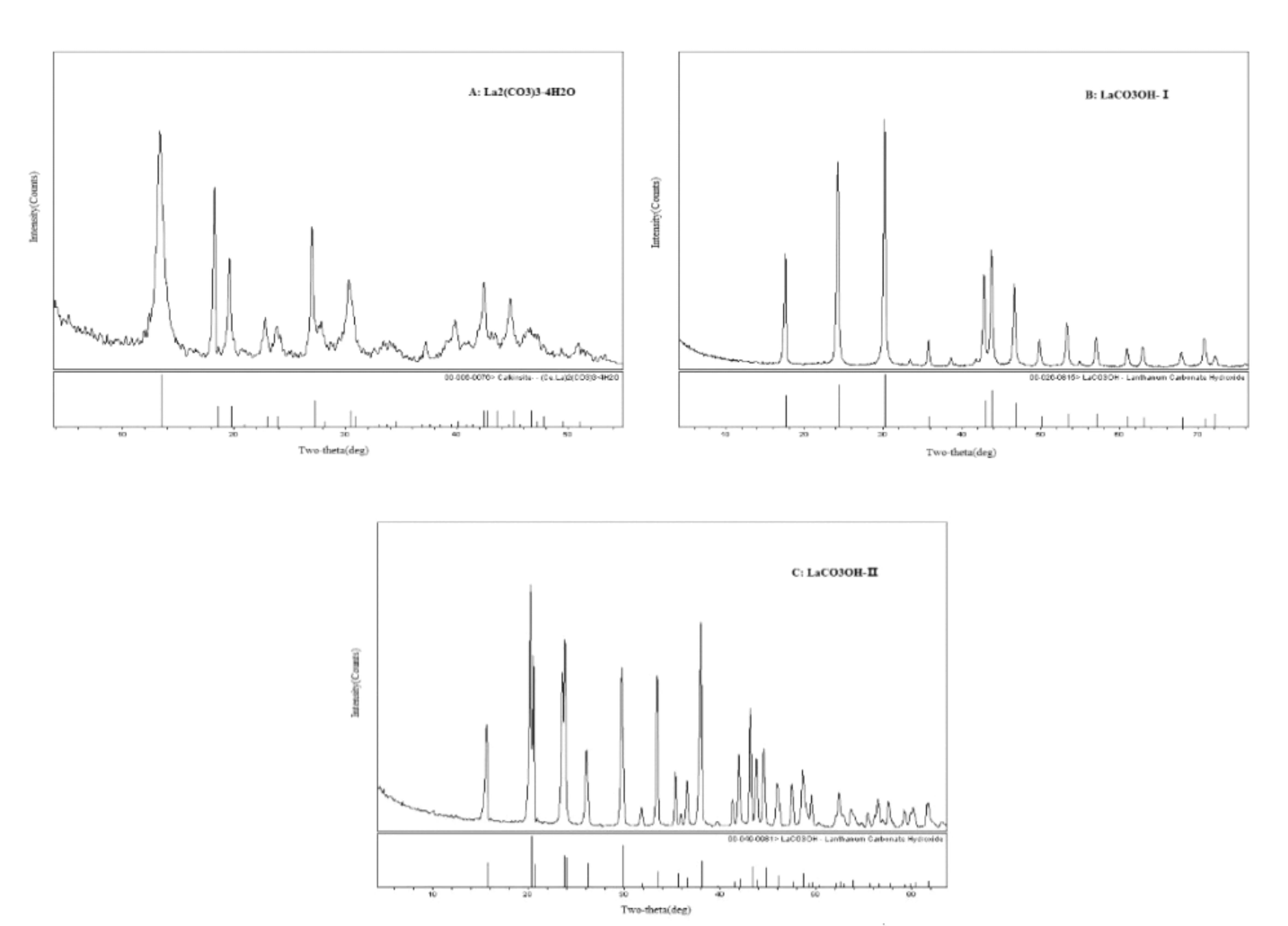
图4 四水碳酸镧、碱式碳酸镧晶型I和II的X射线衍射图A. 四水碳酸镧; B.碱式碳酸镧晶型I ; C. 碱式碳酸镧晶型II
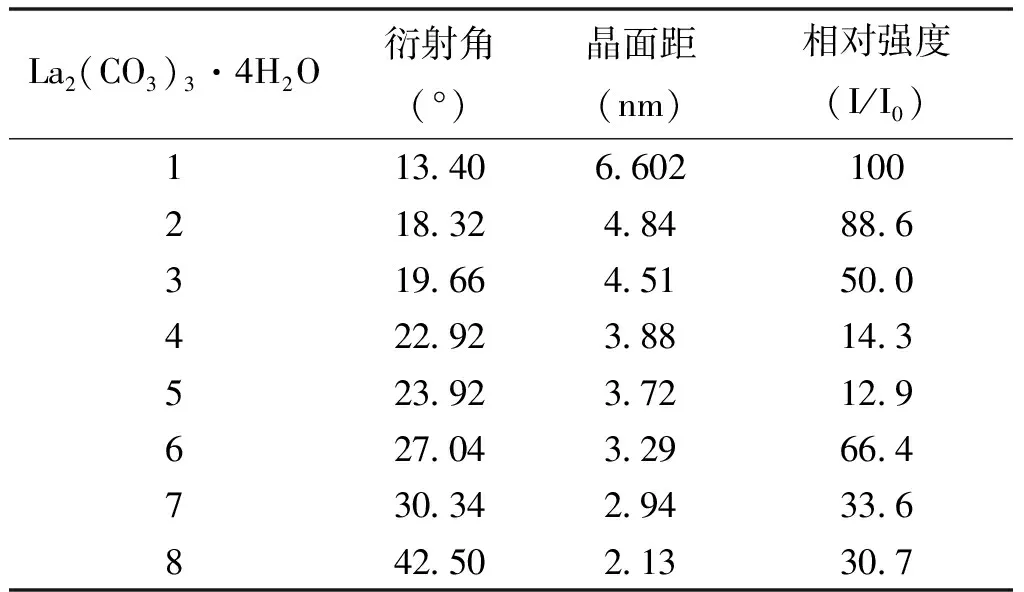
表2 四水碳酸镧的PXRD数据

表3 碱式碳酸镧晶型I的PXRD数据
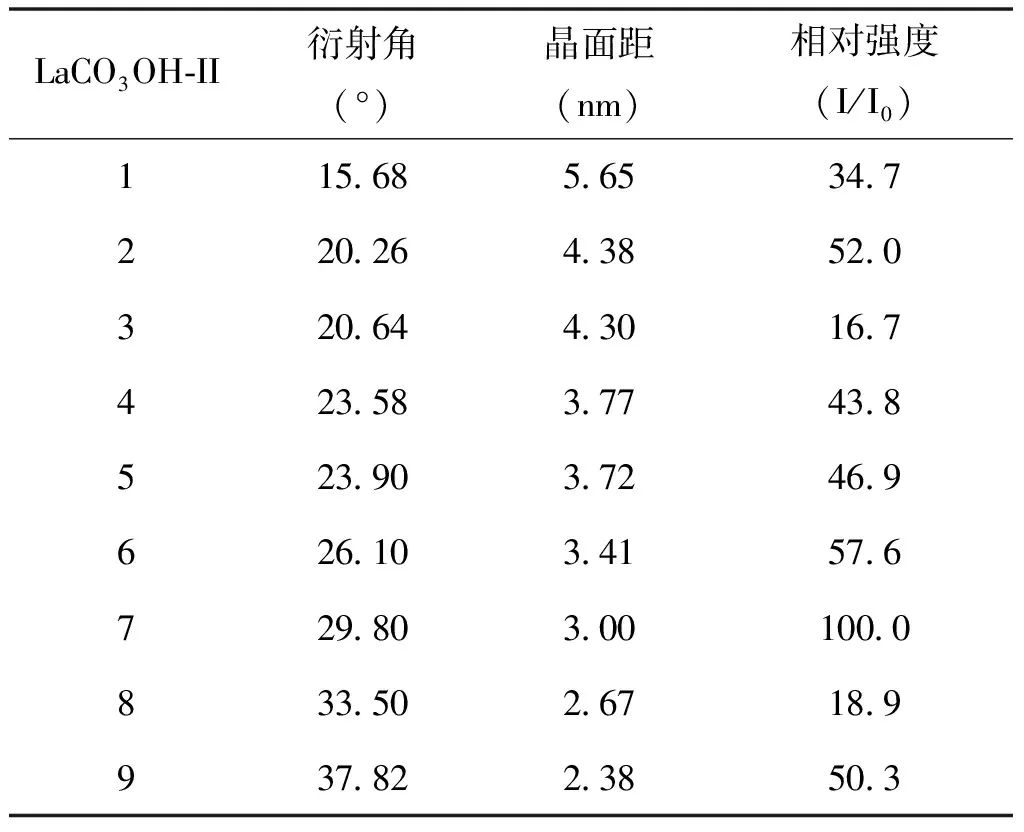
表4 碱式碳酸镧晶型II的PXRD数据
3.2 定量分析
将制备好的碳酸镧和LaCO3OH-I和II样品分别过200目筛,使粒径尽量保持一致便于充分混合减少粒径所引起的实验误差。然后将样品放置于干燥器中平衡24小时。通常采用研磨混合的方法来对样品进行混合。在混合前,采用干法分别对碳酸镧和两种碱式碳酸镧晶型研磨5min、10min和20min,用X射线衍射法对其进行定性检测,结果表明,3种晶型在不同时间研磨后均未发生晶型和物相变化,可以此作为定量分析的基础标样开展后续实验。
在本实验中,首选干法研磨混合的方法来配制用于定量分析的二元混合物实验样品,按照表5中所示比例(总量300mg)分别于分析天平上称取LaCO3OH-I、II型和La2(CO3)3·4H2O样品,配制不同比例混晶体系。再置于玛瑙研钵中研磨混合20min,分别编号N1~N15和M1~M15。将配好的混合样品静置在干燥器中,备用。
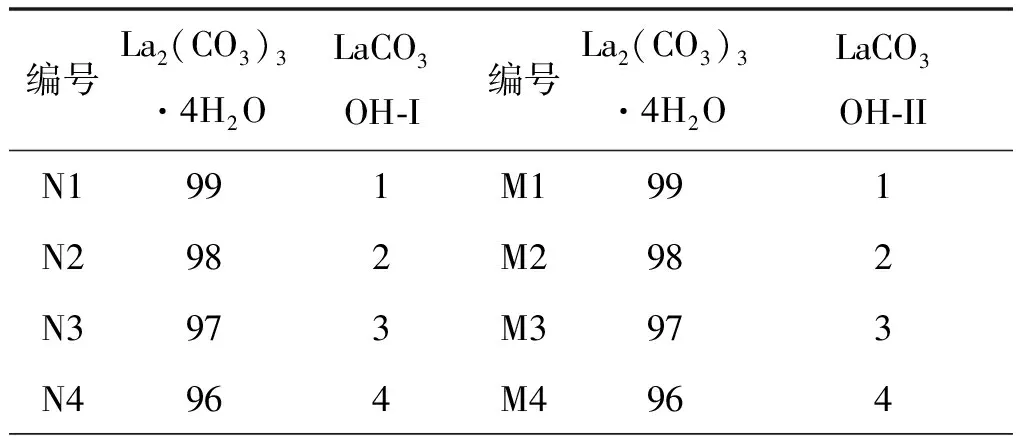
表5 不同比例的碳酸镧与碱式碳酸镧的二元混合体系 %
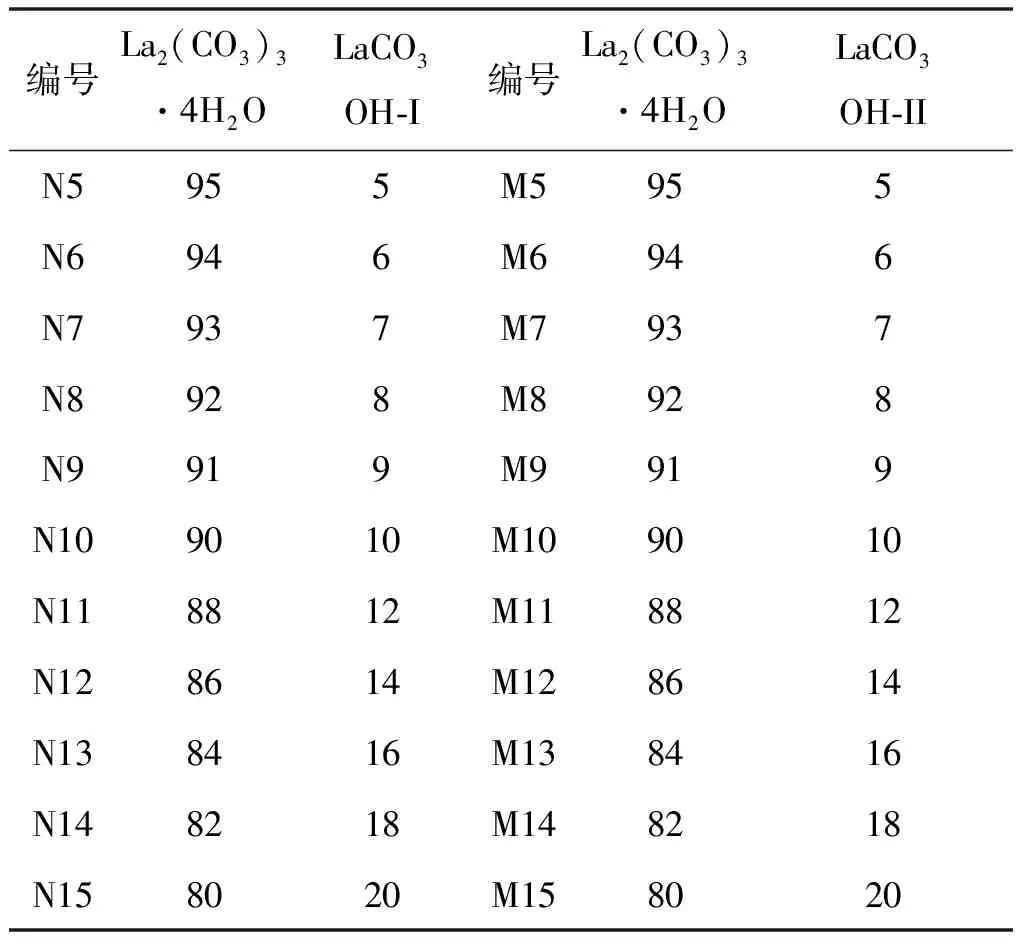
续表5
由上面的定性分析可知,La2(CO3)3·4H2O的特征峰为13.40°,LaCO3OH-I的特征峰为17.62°,LaCO3OH-II的特征峰为26.10°。将按表5制备好的混合样品(N1~N15,M1~M15)在相同的实验条件下进行测定。比例相同的样品制样3次,测定3次,取3次测试结果的平均值作为最终结果。对测试得到的结果分别采用峰高法和峰面积法进行积分,结果如图5和图6所示。杂质LaCO3OH-I的峰高标准曲线方程(A)为y = 0.35865x + 0.03026 ,峰面积标准曲线方程(B)为y= 0.12872x+ 0.0072,R2分别为0.95163和0.98431。杂质LaCO3OH-II的峰高标准曲线方程(A)为y= 0.48405x+ 0.01432 ,峰面积标准曲线方程(B)为y= 0.12774x+ 0.00724,R2分别为0.98661和0.98933。由此可得出结论:两种杂质的定量分析中,峰面积法的线性关系均优于峰高法。
碱式碳酸镧Ⅰ和Ⅱ的检测限和定量限如表6所示,其中采用峰高法计算出碱式碳酸镧Ⅰ和Ⅱ的检测限和定量限分别为3.06%、9.28%和1.32%、4.01%,峰面积法计算出碱式碳酸镧Ⅰ和Ⅱ的检测限和定量限分别为1.71%、5.19%和1.18%、3.58%,由此可以看出,建立两种杂质晶型的标准曲线都是峰面积法更为灵敏。
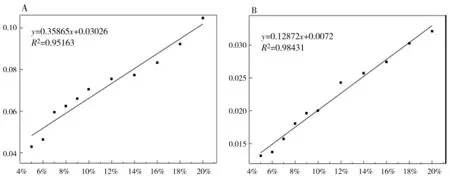
图5 LaCO3OH-I与 La2(CO3)3·4H2O二元混合物的强度比与含量的关系曲线A.峰高法; B. 峰面积法
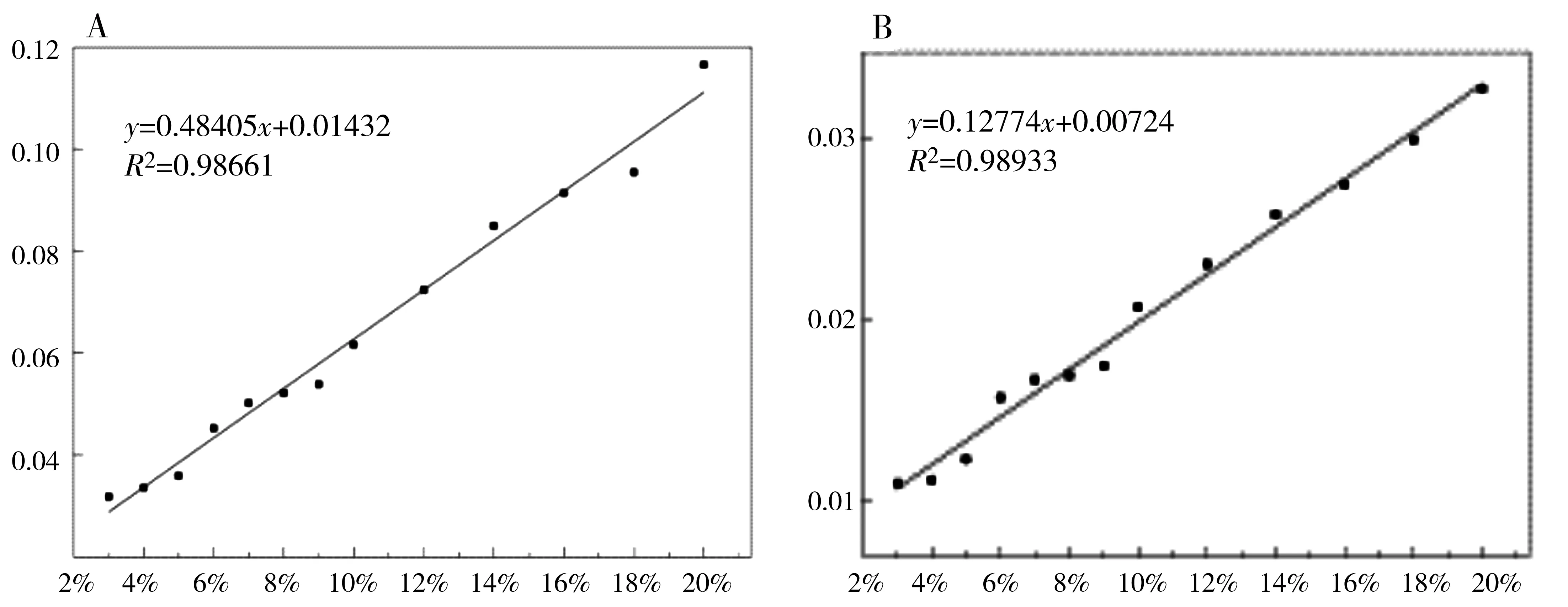
图6 LaCO3OH- II与 La2(CO3)3·4H2O二元混合物的强度比与含量的关系曲线A.峰高法; B.峰面积法

表6 碱式碳酸镧晶型Ⅰ和Ⅱ的检测限和定量限
为了验证标准曲线的准确度,配置了不同比例的LaCO3OH-I、LaCO3OH-II与四水碳酸镧的混合样品(7%、10%和16%),在相同的实验条件下进行测定,将所得数据按同样的方法计算出强度比,再分别代入相应的标准曲线方程,结果如表7所示。采用峰高法作出的LaCO3OH晶型I的标准曲线准确度在77.82%~84.13%,峰面积法的准确度在87.09%~94.51%;采用峰高法作出的LaCO3OH晶型II的标准曲线准确度在83.59%~91.36%,峰面积法的准确度在88.35%~97.37%。由此可见,两种晶型均为峰面积法得出的标准曲线准确度高于峰高法。
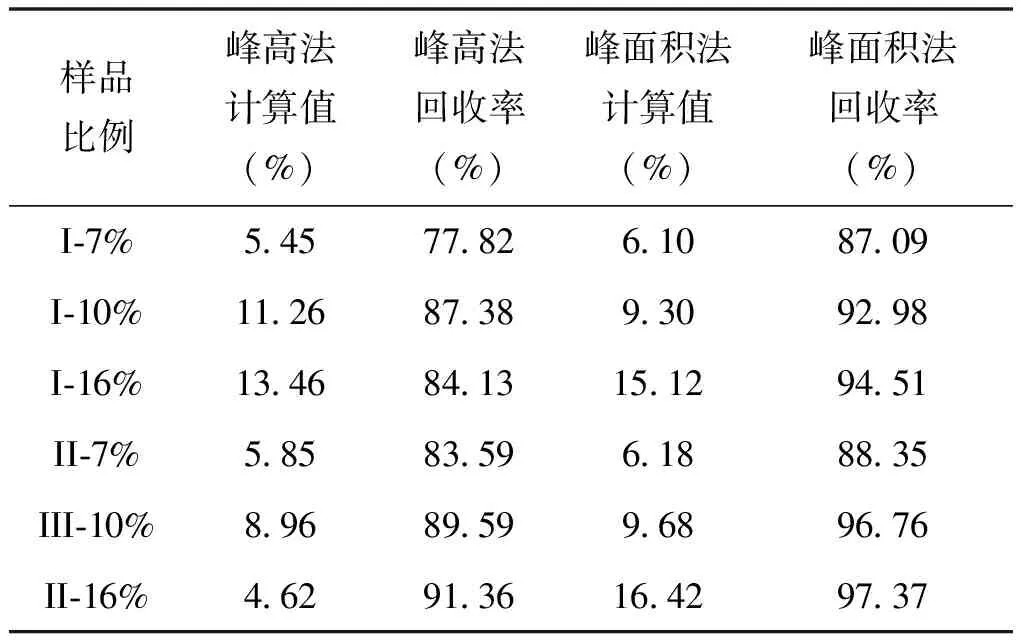
表7 不同比例的碱式碳酸镧晶型I和II的准确度计算值
4 讨论
近年来,药物的晶型研究已经成为国内药物开发和原研药仿制的重要内容之一,同种药物可能存在着不同的晶型,导致药物疗效和生物利用度的不同。对药物晶型进行结构表征成为临床用药的前提,也是药物稳定性考察的基础和依据。因此,在对药物药理作用研究前必须建立一系列系统的表征方法,以确保该生产药物是目标药物。本实验主要通过热重分析(TGA)、傅里叶变换红外光谱分析(FTIR)、扫描电子显微分析(SEM)和X射线粉末衍射分析(PXRD)等现代分析手段对样品进行表征,为药物的定量分析提供参考依据。
本实验在对四水碳酸镧及其两种杂质晶型碱式碳酸镧定性分析的基础上,建立了基于PXRD单峰法中峰高法和峰面积法的定量分析方法。通过选取3种晶型的特征峰来计算峰高强度比和峰面积强度比,分别得出了两种碱式碳酸镧杂质在目标晶型中含量的定量标准曲线,两者都有良好的线性关系,且峰面积法优于峰高法。在X射线衍射的定量分析中,通常每个物相我们选取一条特征衍射峰来采集数据。该衍射峰和其它物相的衍射峰不可发生重叠,尽可能选择独立且强度较高的特征峰,另外制样时还要避免择优取向等原因造成强度不稳定、出现偏差的现象。本实验采用的是干法研磨的手段,在对二元体系进行研磨混合时,力度要适中,重在混合而非用力研磨,否则会导致样品粘附在玛瑙研钵壁或者研磨棒上,造成数据不准确,偏离真实值,甚至破坏晶型。由于LaCO3OH-I和LaCO3OH-II的含量较低,应一边研磨混合一边少量多次加入到La2(CO3)3·4H2O中,这样可以避免杂质样品结块,集中在同一区域从而造成较大实验误差。
猜你喜欢
滇池(2022年5期)2022-04-30 21:44:36
河南水利与南水北调(2021年7期)2021-01-07 05:37:30
中国粉体技术(2021年1期)2021-01-04 02:19:04
河南水利年鉴(2020年0期)2020-06-09 05:43:42
西安工程大学学报(2016年3期)2016-06-05 09:26:35
计量学报(2015年3期)2015-08-10 10:10:19
无机化学学报(2014年7期)2014-02-28 17:32:10
湖南水利水电(2014年2期)2014-02-27 14:45:38
无机盐工业(2012年11期)2012-03-19 21:37:01
环球时报(2012-02-03)2012-02-03 11:04:46
