
纺锤体动粒相关复合体-1(SKA1)促进葡萄膜黑色素瘤细胞增殖的分子机制研究△
2021-08-30 03:08凌峰张勇辛向阳
眼科新进展 2021年8期
凌峰 张勇 辛向阳
葡萄膜黑色素瘤(UM)是成人最常见的原发性眼内恶性肿瘤,恶性程度高、易转移、预后差[1-2]。UM患者尽管以手术为主的综合治疗局部控制良好,但复发、转移仍是术后面临的主要问题[3],目前转移后治疗尚无一致意见,且效果差。针对癌基因设计靶向药物是治疗肿瘤的重要途径之一,因此,深入研究UM发生发展的分子机制、寻找新型肿瘤转移标志物及治疗靶点具有重要的科学意义和临床价值。
纺锤体动粒相关复合体-1(SKA1)是纺锤体和动粒相关复合物(SKA)的重要组成部分(或称亚基),参与有丝分裂的调控。破坏SKA1的灵活性,或其微管蛋白结合位点,都能扰乱有丝分裂的正常进程,使有丝分裂阻滞于分裂中期,无法进入后期[4-5]。有文献报道,SKA1在胃癌、肝细胞癌、非小细胞肺癌等恶性肿瘤发生发展过程中发挥重要作用[6-7],然而,SKA1在UM组织及细胞中的表达、作用及分子机制目前尚不清楚。本研究首次探讨了SKA1在UM发生发展过程中的作用,并通过基因表达谱芯片联合生物信息学分析探寻SKA1的分子机制。
1 材料与方法
1.1 细胞株和细胞培养A-375、MUM-2B和MUM-2C细胞株由天津医科大学眼科医院孙丰源教授惠赠。细胞培养使用含体积分数10%胎牛血清(FBS)的高糖DMEM培养液(美国Gibco公司),并加入10 g·L-1青链霉素预防细菌污染,置于37 ℃、含体积分数5% CO2培养箱中,间隔2~3 d更换培养基。
1.2 筛选SKA1敲减的稳转细胞株重悬A-375、MUM-2B和MUM-2C细胞,细胞浓度均调整为4×104个·mL-1,各吸取2 mL细胞悬液,接种于6孔板,继续培养,细胞汇合度达30%~40%时,吸去培养液,以2 mL含体积分数10%FBS的培养液稀释慢病毒(购自上海吉凯),包括靶向SKA1敲减慢病毒[shSKA1;敲减靶点序列分别为5’-GGAGATGAGATCATTGTAA-3’(shSKA1-1)、5’-GGATACCAAAGGTCGTTATTT-3’(shSKA1-2)、5’-CAATGCTGCAGACCCTAATAA-3’(shSKA1-3)]以及空载体慢病毒(shCtrl),加入助转剂聚凝胺(Polybrene,2 g·L-1),混匀后加入相应孔中。感染慢病毒48~72 h后,换含嘌呤霉素(2 g·L-1)的培养基进行抗性筛选,计算细胞存活率即阳性感染率,最终筛选出SKA1敲减的稳转细胞株作为SKA1敲减组用于后续实验。
1.3 实时荧光定量PCR、Western blot检测使用Trizol试剂分别提取A-375、MUM-2B和MUM-2C细胞总RNA,依据说明书逆转录成cDNA,实时荧光定量PCR(qRT-PCR)检测,反应体系:SYBR premixex Taq 6.0 μL、上下游引物各0.5 μL、cDNA模板1.0 μL、RNase-Free H2O 4.0 μL,以GAPDH 作为内参,SKA1相对表达量通过 2-△△Ct或ΔCt计算获得。
RIPA细胞裂解液提取SKA1敲减组、对照组(转染shCtrl)的MUM-2B细胞总蛋白,以BCA试剂盒测定蛋白浓度,取30 μg总蛋白上样于SDS-PAGE凝胶,电泳后电转至PVDF膜,分别与SKA1抗体(11000,北京博奥森)和GAPDH(1500,Santa Cruz Biotechnology公司,美国)4 ℃孵育过夜,漂洗后与HRP标记的二抗(12000,Santa Cruz Biotechnology公司,美国)室温孵育2 h,化学发光试剂显影,然后采用Quantity one扫描软件分析。
1.4 MTT检测细胞增殖取对数生长期的SKA1敲减组和对照组MUM-2B细胞,于96孔板中按每孔3000个细胞铺板,在培养后不同时间点(1 d、2 d、3 d、4 d、5 d)加入20 μL 5 g·L-1的MTT试剂于孔中,反应4 h,持续振荡6 min,酶标仪检测490 nm/570 nm处光密度。
1.5 流式细胞术检测细胞凋亡慢病毒转染MUM-2B细胞48 h后,用2.5 g·L-1胰蛋白酶消化离心SKA1敲减组和对照组MUM-2B细胞,制成单细胞悬液,细胞浓度为2×104个·mL-1。800 r·min-1离心5 min,弃上清,加5 mL PBS重悬细胞,重复2次后离心,弃上清,最后重悬细胞于0.5 mL PBS中。用低速振荡器边振荡边加入5 mL预冷的含体积分数70%乙醇,4 ℃过夜。第二日将固定好的细胞以1000 r·min-1离心5 min,弃上清,PBS清洗后重悬细胞。加入5 μL(10 g·L-1)的RNaseA,37 ℃消化1 h,加入终浓度50 g·L-1碘化丙啶,4 ℃避光染色过夜,使用流式细胞仪分析软件Guava InCyte分析。
1.6 Caspase-3/7法检测细胞凋亡SKA1敲减组、对照组细胞计数后,按每孔1×104个细胞加入96孔板,细胞继续培养2 d,每孔中加入Caspase-Glo反应液100 μL,500 r·min-1离心30 min,再室温孵育 1 h,酶标仪检测仪器测定信号强度。
1.7 基因表达谱芯片分析提取SKA1敲减组和对照组MUB-2B细胞的RNA,反转录成cDNA,进行表达谱芯片检测。再采用R limma包筛选差异表达基因,并对差异基因进行GO(Gene Ontology)和KEGG (Kyoto Encyclopedia of Genes and Genomes)富集分析,初步探索SKA1促进UM细胞增殖的分子机制。
1.8 SKA1生存分析从TCGA GDC网站(https://portal.gdc.cancer.gov/)分别下载UM转录组、临床数据文件,利用定制化perl脚本将下载的UM样本文件整理成基因转录组矩阵文件、临床文件;再将转录组、临床文件合并,采用R“survminer”和“survival”包,绘制Kaplan-Meier生存曲线图。依据SKA1表达中位值将UM样本分为SKA1高、低表达组,利用基因集富集分析(Gene set enrichment analysis)软件,解析与SKA1表达水平与各信号通路状态(激活、抑制或无变化)的关系。

2 结果
2.1 SKA1敲减序列的筛选以qRT-PCR检测SKA1在不同黑色素瘤细胞株中的表达,相对表达量以-ΔCt表示,结果显示,SKA1在A-375、MUM-2B和MUM-2C细胞株的表达丰度均高(图1A),但在细胞培养过程中,A-375、MUM-2C生长状态差,经检验为支原体污染,遂采用MUM-2B细胞用于后续实验。qRT-PCR检测MUM-2B各处理组SKA1表达,结果显示,与shCtrl组相比,SKA1敲减组表达显著降低(P<0.01),shSKA1-1、shSKA1-2、shSKA1-3各敲减组的敲减效率分别为78.86%、62.71%、57.83%。因此,shSKA1-1用于后续细胞表型功能实验(图1B)。

图1 SKA1在三种细胞株的表达丰度及靶向SKA1敲减序列的筛选 A:SKA1在三种细胞株中的表达丰度;B:SKA1不同敲减靶点的敲减率(**P<0.01)。
2.2 SKA1对MUM-2B细胞增殖的影响MTT检测结果显示,对照组MUM-2B细胞培养1 d、2 d、3 d、4 d、5 d时光密度分别为0.20±0.00、0.36±0.01、0.61±0.02、0.98±0.02、1.25±0.03;SKA1敲减组MUM-2B细胞培养1 d、2 d、3 d、4 d、5 d 时光密度分别为0.15±0.00、0.20±0.04、0.25±0.02、0.37±0.01、0.41±0.05;与对照组相比,SKA1敲减组MUM-2B细胞培养3 d、4 d、5 d 的光密度均显著降低(均为P<0.01),说明SKA1敲减后MUM-2B细胞增殖活性被显著抑制(图2)。

图2 对照组、SKA1敲减组MUM-2B细胞MTT生长曲线 注:与对照组比较,**P<0.01,***P<0.001。
2.3 SKA1对MUM-2B细胞凋亡的影响流式细胞仪检测结果显示,对照组细胞凋亡率为5.30%±0.45%,SKA1敲减组细胞凋亡率为12.63%±0.04%,SKA1敲减组细胞凋亡率显著高于对照组(P<0.01)。采用Caspase-3/7法检测Caspase-3/7活性,结果显示,与对照组相比,SKA1敲减组细胞Caspase-3/7活性显著增强(P<0.001),与流式细胞仪检测的细胞凋亡情况一致(图3)。
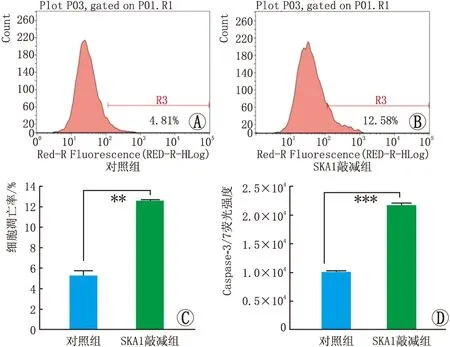
图3 对照组、SKA1敲减组MUM-2B细胞凋亡比例图 A:对照组细胞流式凋亡图;B:SKA1敲减组细胞流式凋亡图;C:两组细胞凋亡统计图;D:两组细胞Caspase-3/7荧光强度统计图。注:**P<0.01,***P<0.001。
2.4 SKA1促进UM细胞增殖的分子机制分别提取对照组和SKA1敲减组MUM-2B细胞的RNA,反转录成cDNA,进行基因表达谱芯片检测(图4)。采用R“limma”包筛选差异表达基因,共筛选出差异表达基因362个,其中高表达176个,低表达186个,高、低表达组分别选取差异最显著的前15个基因,以热图展示(图4A);并对所有基因及后续富集通路中的基因以火山图展示(图4B)。经GO和KEGG通路富集分析显示,362个差异基因显著富集于P53

图4 SKA1敲减组、对照组全基因表达谱测序热图、火山图、富集分析柱状图以及相应细胞Western blot图 A:高、低表达组中差异最显著的前15个基因的热图; B:全部基因包含P53信号通路8个差异基因的火山图; C:差异基因GO柱状图; D:差异基因KEGG富集分析柱状图;E:各组IGFBP-3表达Western blot图。
信号通路(图4C、图4D),富集在此通路的基因包括胰岛素样生子因子结合蛋白-3(IGFBP3)、SESN2、TNFRSF10B、CCNG1、CD82、ATR、CCNE、RCHY1。为验证生物信息学的分析结果,分别提取SKA1敲减组及对照组细胞总蛋白,Western blot检测结果显示(图4E),SKA1敲减组IGFBP3表达下调,提示SKA1通过负调控IGFBP3抑制P53信号通路,从而促进UM细胞的增殖。
2.5 SKA1的高、低相对表达对UM患者生存期的影响将TCGA数据库UM样本按SKA1表达(FPKM)最佳界值(Best cutoff=0.64)分为高、低表达患者,绘制Kaplan-Meier生存曲线,结果显示,SKA1高表达患者生存率显著下降(P<0.05;HR=2.55,95%CI:0.92~7.05)(图5)。

图5 SKA1的不同表达水平在UM患者中Kaplan-Meier生存曲线图
3 讨论
UM是成人最常见的眼内原发性恶性肿瘤,对于高危患者,可采用的有效治疗方案不足1%[8],手术患者约50%以上出现血行转移,大部分累及肝脏,最终致肝衰竭而死亡。手术切除既没明显改善患者的生活质量,又没达到根治肿瘤的效果。因此,深入研究UM发生及转移的分子机制、寻找肿瘤分子标志物及治疗新靶点具有重要意义和临床应用价值。
有文献报道,SKA1参与胃癌、肝细胞癌、口腔鳞癌、涎腺腺样囊性癌、非小细胞肺癌、膀胱癌、前列腺癌、神经胶质瘤、甲状腺乳头状癌、骨肉瘤等恶性肿瘤的转移[9-14]。然而,SKA1在UM发生发展过程中的作用及分子机制目前尚不清楚。本研究首次发现SKA1通过P53/IGFBP3促进UM细胞增殖,抑制其凋亡,具有潜在的临床应用价值。
SKA1介导的信号通路在不同肿瘤中影响不同表型。Li等[15]通过体外实验发现,SKA1除了在有丝分裂中富集于纺锤体微管和动粒外层,也会在培养细胞的中心体富集。在SKA1转基因裸鼠中,敲除SKA1导致中心体复制障碍,而SKA1过表达导致前列腺上皮细胞的中心体过度扩增,出现多个中心体,导致裸鼠的肿瘤发生概率增高。Zhang等[16]发现,SKA1敲减后,口腔鳞状细胞癌CAL-27细胞的增殖、克隆形成能力下降,凋亡增加,细胞周期被阻滞于G2/M期。Shi等[17]研究发现,SKA1敲减后,A172、U251神经胶质瘤细胞增殖、侵袭能力减弱等。为进一步研究SKA1在UM的发生发展中是否发挥了促进细胞增殖、抑制细胞凋亡的作用,我们利用qRT-PCR 检测三种UM细胞株中SKA1表达,并筛选出最有效的敲减序列shSKA1,用于后续的MTT实验及流式细胞技术、Caspase-3/7法检测。结果发现,敲减了SKA1的MUM-2B细胞增殖活性显著低于对照组,而细胞凋亡速度显著高于对照组。间接证明了SKA1在UM的发生发展中发挥促进增殖、抑制凋亡的作用,这与其他文献[16-17]所报道结果相一致。
IGFBP3是胰岛素样生长因子(IGFs)系统中的六种高亲和力蛋白IGFBPs中作用最主要的调节蛋白,其主要功能是结合循环中的IGFs,从而控制它们的生物利用率[18]。研究发现,IGFBP3可以通过控制IGFs对胰岛素样生长因子受体(IGF1R)的作用,从而发挥其抑制增殖和抗凋亡的作用[19]。Yang等[20]在体内外实验中均证实,IGFBP3的过表达均能抑制胶质细胞瘤肿瘤细胞增殖,进而抑制肿瘤生长。有研究表明,IGFBP3是P53调控的靶基因,其表达随野生型P53的增加而增加,并与IGFBP3启动子中的DNA反应元件结合[21]。P53诱导IGFBP3表达已被证明与细胞凋亡增加和增殖减少有关[22]。这些都证明 IGFBP3 蛋白在肿瘤发生发展过程中发挥抑癌基因的作用。为探索SKA1影响UM的潜在通路,我们利用KEGG富集分析发现,SKA1可能通过P53信号通路发挥作用, Western blot检测结果显示,SKA1敲减后P53通路中关键分子IGFBP3表达随之下降,提示SKA1可能通过P53/IGFBP3 来影响UM细胞的增殖及凋亡。
同时,我们还分析了TCGA数据库UM样本中SKA1表达与临床及预后的相关性。由于TCGA数据库没有UM癌旁正常组织,我们分析SKA1的表达差异,只能分析其与临床及预后的关系。结果显示,SKA1高表达的患者总生存率明显降低,这与Dong等[23]提出的SKA1是影响甲状腺乳头状癌预后的独立危险因素一致。
综上所述,本研究发现SKA1是影响UM预后的独立危险因素,通过P53/IGFBP3信号通路促进UM细胞增殖,抑制细胞凋亡。因此,SKA1增殖可能成为UM分子治疗的潜在靶点,然而,SKA1直接作用机制,包括与SKA1结合的上、下游蛋白分子及功能域尚不明确,有待进一步研究。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
检验医学与临床(2022年19期)2022-10-10
中国现代医生(2022年19期)2022-08-25
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
中国现代医生(2018年22期)2018-12-04
中国报道(2018年2期)2018-04-20
