
CuO/CeO2催化剂的制备及CO氧化性能研究
2021-08-26 09:03:22朱明路张煜华李金林
化学与生物工程 2021年8期
朱明路,张煜华,李金林
(中南民族大学化学与材料科学学院,湖北 武汉 430074)
一氧化碳(CO)是工业生产、工业制造和冶金的副产物,工业CO排放多为固定源,且排放量较少[1-3];而以化石燃料作为动力的汽车、火车、轮船、飞机等移动源才是CO排放的主要来源,其中汽车的CO排放量占90%左右,道路交通是CO的主要来源[4-7],因此,有必要对汽车尾气进行减排。减少汽车尾气排放主要有3种途径:改进汽车技术和改造发动机[8]、寻找新型清洁能源[9]、使用催化转化器[10],其中催化转化器应用最广。近年来,美国能源部提出“150 ℃挑战”,即开发在150 ℃以下可将标准污染物消除90%以上的催化系统[11]。我国汽车排放标准也逐步提高,开发新型尾气处理催化剂一直是近年来的研究热点。当前,汽车尾气处理催化剂主要以铂族贵金属作为活性成分,由于Pt、Pd、Rh等价格昂贵,储量较少,高温下易烧结[12-14],所以开发少用或不用贵金属的汽车尾气处理催化剂具有现实意义。研究发现,CuO/Al2O3催化剂具有CO氧化活性,但催化活性并不理想[15],而CuO/CeO2催化剂由于CuO与CeO2间的金属载体相互作用,表现出良好的CO氧化性能,温度是影响CuO/CeO2催化性能的重要因素[16]。基于此,作者在不同焙烧温度下制备CuO/Al2O3和CuO/CeO2催化剂,通过CO氧化反应,并结合BET、XRD、SEM、EDS、H2-TPR、Raman和XPS等表征,探究CuO/CeO2催化剂具有高CO氧化活性的原因。
1 实验
1.1 催化剂的制备
1.1.1 Al2O3及CeO2载体的预处理
称取一定量的Al2O3置于坩埚中,再将坩埚放入马弗炉中,以2 ℃·min-1的升温速率从室温升至550 ℃,保持5 h,得到焙烧处理后的Al2O3载体,命名为Al2O3-550。
称取一定量的Ce(NO3)3·6H2O置于坩埚中,再将坩埚放入马弗炉中,以2 ℃·min-1的升温速率从室温升至350 ℃,保持2 h,得到焙烧处理后的CeO2载体,命名为CeO2-350。
1.1.2 CuO/Al2O3及CuO/CeO2催化剂的制备
称取0.81 g Cu(NO3)2·3H2O,加入7.89 g(以0.5 g Al2O3-550进行水饱和计算得出)去离子水,超声5 min使Cu(NO3)2分散均匀;再称取4 g Al2O3-550载体置于研钵中,用滴管将Cu(NO3)2溶液滴入研钵,充分研磨,多次重复该步骤,直至将Cu(NO3)2溶液滴完为止;于120 ℃烘箱中干燥6 h,除去水分;将干燥好的样品分为2份,置于马弗炉中,以2 ℃·min-1的升温速率从室温分别升至400 ℃和600 ℃,保持6 h,即得CuO/Al2O3催化剂,分别命名为CuO/Al2O3-400和CuO/Al2O3-600。
称取0.81 g Cu(NO3)2·3H2O,加入2.10 g(以0.5 g CeO2-350进行水饱和计算得出)去离子水,同法制备CuO/CeO2催化剂,分别命名为CuO/CeO2-400和CuO/CeO2-600。
1.2 催化剂的表征
BET分析:采用Micromeritics ASAP 2460型比表面积和孔隙度分析仪测定催化剂的比表面积。将催化剂粉末放入石英管中,升温至200 ℃并保持6 h,再用液氮冷却至-196 ℃,测定催化剂的比表面积。
XRD分析:采用D8 Advance型X-射线衍射仪分析催化剂的物相组成。Cu κα辐射源(λ=0.1506 nm),工作电压40 kV,工作电流40 mA,2θ范围10°~80°。
SEM及EDS分析:采用Talos F200X型透射电子显微镜分析催化剂的形貌和元素分布。用无水乙醇分散催化剂粉末并超声10 min,取悬浮液滴于钼网上,干燥后测试,电子枪加速电压20~200 kV。
H2-TPR分析:采用AMI-300型多功能表征仪分析催化剂的还原能力。称取30 mg催化剂粉末放入U型石英管中,在Ar气氛下将床层温度升至120 ℃并吹扫1 h;待床层温度降至70 ℃,切换为5% H2/Ar(流速30 mL·min-1)混合气吹扫至TCD基线平稳后,将床层温度以10 ℃·min-1的升温速率从70 ℃升至750 ℃,并保持30 min。
Raman光谱分析:采用DXR2 xi型显微拉曼高速成像光谱仪分析催化剂的结构。激发波长532 nm,光谱范围50~3 500 cm-1。
XPS分析:采用VG Multilab 2000型光电子能谱仪分析催化剂表面的元素价态、相对含量等。以Alκα射线作为激发源,以催化剂表面污染碳的C1s峰结合能(284.8 eV)作为内标校正。
1.3 催化剂的CO氧化性能测试
催化剂的CO氧化性能测试在6230型固定床反应器(北京拓川)上进行。称取0.037 5 g 40~60目的催化剂,加入0.375 g石英砂,混匀后装填于石英反应管中;300 ℃下通入20% O2/Ar(流速125.0 mL·min-1)将催化剂预处理1 h,自然冷却至室温;通入5% CO/Ar(流速50.0 mL·min-1)、20% O2/Ar(流速62.5 mL·min-1),用纯Ar(流速12.5 mL·min-1)进行平衡,总流速125.0 mL·min-1,空速200 000 mL·g-1·h-1;将反应器以2 ℃·min-1的升温速率从室温升至600 ℃,在反应器排气口接入Agilent GC 7890B型气相色谱仪对气体组成进行在线分析。按下式计算CO转化率(XCO,%):
式中:nCO,in为入口气流中CO的含量;nCO,out为出口气流中CO的含量。
2 结果与讨论
2.1 BET分析
为了探究不同焙烧温度下催化剂的稳定性,对Al2O3-550载体、CuO/Al2O3催化剂、CeO2-350载体、CuO/CeO2催化剂的比表面积进行测定,结果如图1所示。

图1 载体和催化剂的BET比表面积Fig.1 BET specific surface areas of supports and catalysts
由图1可知,(1)Al2O3-550载体、CuO/Al2O3-400催化剂、CuO/Al2O3-600催化剂的比表面积分别为177.3 m2·g-1、160.7 m2·g-1、152.1 m2·g-1。CuO/Al2O3催化剂的比表面积较Al2O3-550载体出现了一定程度的减小,这是由于,CuO占据了Al2O3-550载体的部分孔道,造成比表面积减小。此外,焙烧温度升高,造成CuO颗粒长大,占据Al2O3-550载体更多的孔道,导致CuO/Al2O3-600催化剂的比表面积较CuO/Al2O3-400催化剂的进一步减小。(2)CeO2-350载体、CuO/CeO2-400催化剂、CuO/CeO2-600催化剂的比表面积分别为58.7 m2·g-1、55.3 m2·g-1、55.3 m2·g-1。CuO/CeO2催化剂的比表面积与CeO2-350载体相比,基本能够得到保持,CuO/CeO2-600催化剂与CuO/CeO2-400催化剂的比表面积保持一致,这是由于,CuO与CeO2间存在金属载体相互作用,CuO能够在CeO2-350载体表面高度分散,未出现CuO颗粒长大的现象。
2.2 XRD分析
为了探究载体和催化剂的物相组成,对Al2O3-550载体、CuO/Al2O3催化剂、CeO2-350载体、CuO/CeO2催化剂进行XRD分析,结果如图2所示。
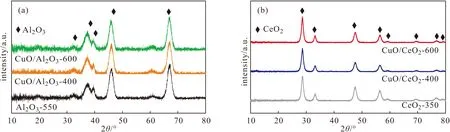
图2 Al2O3-550载体、CuO/Al2O3催化剂(a)及CeO2-350载体、CuO/CeO2催化剂(b)的XRD图谱Fig.2 XRD patterns of Al2O3-550 support,CuO/Al2O3 catalysts(a) and CeO2-350 support,CuO/CeO2 catalysts(b)
由图2可知,CuO/Al2O3催化剂与Al2O3-550载体的特征衍射峰位置基本一致,并未观察到CuO的特征衍射峰;CuO/CeO2催化剂的XRD图谱出现类似情况。这是由于CuO含量较少,能够在Al2O3-550或CeO2-350载体表面高度分散,因此并未观察到CuO的特征衍射峰。
2.3 SEM及EDS分析
为了探究焙烧温度对CuO/Al2O3、CuO/CeO2催化剂的形貌及元素分布的影响,对CuO/Al2O3、CuO/CeO2催化剂进行了SEM和EDS分析,结果如图3所示。

图3 CuO/Al2O3和CuO/CeO2催化剂的SEM照片及EDS图像Fig.3 SEM & EDS images of CuO/Al2O3 and CuO/CeO2 catalysts
由SEM照片可以看出,CuO/Al2O3和CuO/CeO2催化剂均为不规则颗粒状,颗粒大小较为均一;焙烧温度达到600 ℃,催化剂的形貌并未发生明显变化。由EDS图像可以看出,CuO/Al2O3和CuO/CeO2催化剂上Al、Ce和Cu元素都能均匀分散,表明焙烧温度对催化剂的元素分布影响不大。
2.4 H2-TPR分析
为了探究载体和催化剂的还原能力,对Al2O3-550载体、CuO/Al2O3催化剂、CeO2-350载体、CuO/CeO2催化剂进行H2-TPR分析,结果如图4所示。
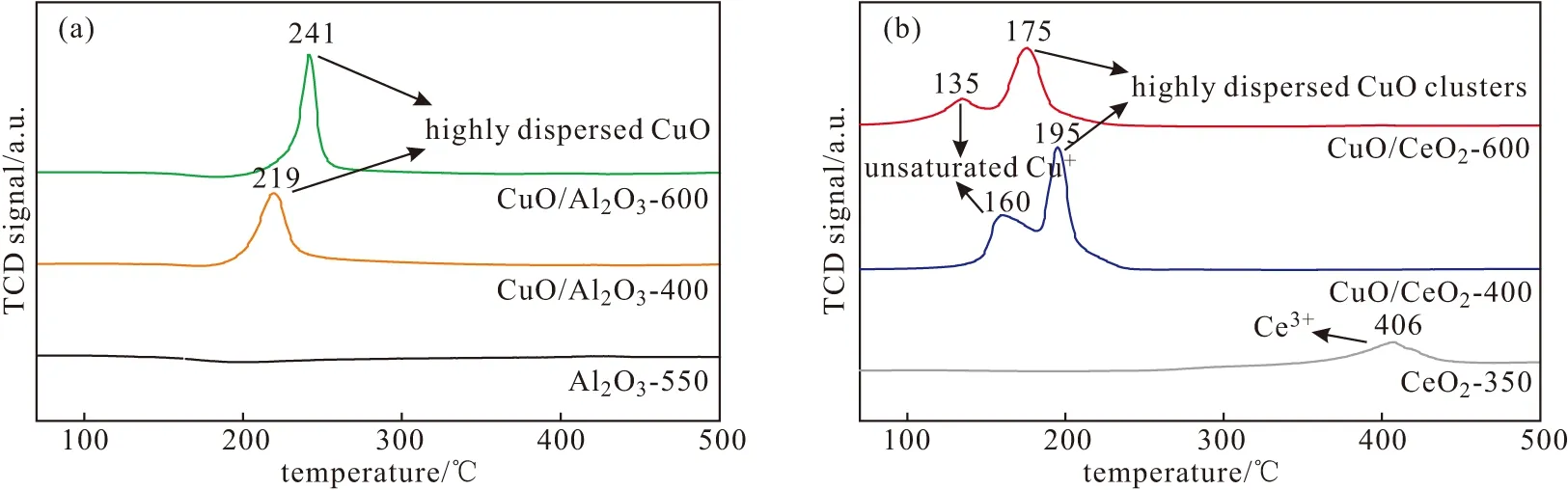
图4 Al2O3-550载体、CuO/Al2O3催化剂(a)及CeO2-350载体、CuO/CeO2催化剂(b)的H2-TPR曲线Fig.4 H2-TPR profiles of Al2O3-550 support,CuO/Al2O3 catalysts(a) and CeO2-350 support,CuO/CeO2 catalysts(b)
由图4a可知,Al2O3-550载体的H2-TPR曲线为一条接近水平的直线,并未观察到明显的还原峰;CuO/Al2O3催化剂的H2-TPR曲线只出现一个还原峰,归属于Al2O3-550载体上高度分散的CuO颗粒的还原;不同温度焙烧的CuO/Al2O3-400和CuO/Al2O3-600催化剂还原峰出现的位置分别为219 ℃和241 ℃,焙烧温度升高,CuO/Al2O3催化剂的还原峰向高温区出现轻微偏移,与文献[15]报道相符,即随着焙烧温度的升高,CuO颗粒在Al2O3-550载体上逐渐长大。
文献[17]报道,CeO2仅在350 ℃以上才开始还原,350 ℃以下的还原峰与CuO物种的还原有关。由图4b可知,CeO2-350载体的H2-TPR曲线在406 ℃出现一个还原峰,归属于CeO2-350载体表面的Ce3+还原;CuO/CeO2催化剂的H2-TPR曲线出现2个近重叠的还原峰,前者归属于与CeO2-350载体紧密接触、在CeO2-350载体上高度分散并与其相互作用形成的配位不饱和的Cu+物种,具有较高的催化活性[18];后者归属于在CeO2-350载体表面高度分散并与其相互作用的CuO团簇的还原[19];焙烧温度升高,CuO/CeO2催化剂的2个还原峰向低温区移动,表明CuO与CeO2间的金属载体相互作用增强,同时,CuO得到更好的分散。与CuO/Al2O3催化剂比较,CuO/CeO2催化剂上与CuO有关的还原峰向低温区移动,这是由于CuO与CeO2间的金属载体相互作用所致;与CuO有关的还原峰位置与CuO/Al2O3、CuO/CeO2催化剂的CO氧化活性相关,CuO与CeO2间的金属载体相互作用越强,CO氧化活性越高。
2.5 Raman光谱分析
为了探究CeO2-350载体和CuO/CeO2催化剂的结构,对CeO2-350载体和CuO/CeO2催化剂进行Raman光谱分析,结果如图5所示。

图5 CeO2-350载体和CuO/CeO2催化剂的Raman光谱Fig.5 Raman spectra of CeO2-350 support and CuO/CeO2 catalysts
由图5可知,CeO2-350载体和CuO/CeO2催化剂在约460 cm-1处均出现一个强的、稍不对称的谱峰,归属于CeO2立方萤石结构的F2g振动模式[20]。文献[21]报道,F2g谱峰的位置受CeO2颗粒大小以及Cu2+掺入CeO2晶格的影响。CeO2-350载体、CuO/CeO2-400催化剂、CuO/CeO2-600催化剂的F2g谱峰分别位于459.1 cm-1、456.5 cm-1、457.6 cm-1处,与CeO2-350载体比较,CuO/CeO2催化剂的F2g谱峰向低波数移动,表明CuO与CeO2间存在金属载体相互作用,部分Cu2+进入到CeO2晶格所致。另外,CeO2-350载体、CuO/CeO2-400催化剂、CuO/CeO2-600催化剂的F2g半峰宽分别为26.3 cm-1、32.1 cm-1、31.5 cm-1,与CeO2-350载体比较,CuO/CeO2催化剂的F2g半峰宽变的更宽,进一步验证了上述结论。CuO/CeO2催化剂在600 cm-1处出现一个较宽的谱峰,而CeO2-350载体在该处并未出现,这是Cu2+进入到CeO2晶格中,造成部分Ce4+转变为Ce3+,为了维持CuO/CeO2催化剂的电荷平衡,同时形成氧缺陷[22]。CuO/CeO2-400和CuO/CeO2-600催化剂在600 cm-1处的谱峰强度基本一致,表明焙烧温度不会影响CuO与CeO2间的金属载体相互作用;260 cm-1出现的一个较弱的谱峰,归属于O原子从理想的萤石结构发生的移动,进一步证明CuO与CeO2间存在金属载体相互作用[23]。
2.6 XPS分析
为了探究载体和催化剂表面的元素价态、相对含量,对CeO2-350载体、CuO/CeO2催化剂进行XPS分析,结果如图6所示,相关数据列于表1。
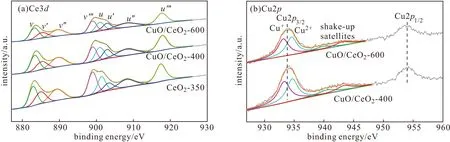
图6 CeO2-350载体和CuO/CeO2催化剂的XPS图谱Fig.6 XPS spectra of CeO2-350 support and CuO/CeO2 catalysts

表1 Ce和Cu的氧化态和表面组成相关数据
由图6可知,Ce3d被分为4对自旋轨道的双峰,分别记为ν和u,对应于Ce3d3/2和Ce3d5/2,其中ν′、u′归属于Ce3+,其余归属于Ce4+[24-25]。相较于CeO2-350载体,CuO/CeO2催化剂的Ce3+含量、氧缺陷Vo含量有所增加(表1),表明CuO与CeO2间存在金属载体相互作用,形成更多的氧缺陷。Cu2p3/2被分为2个峰,前者归属于Cu+,后者归属于Cu2+,并在940~945 eV伴随着卫星峰(Cusatel)。文献[18]报道,只含有Cu2+时,Cusatel/Cu2p3/2值为0.55,CuO/CeO2-400和CuO/CeO2-600催化剂的Cusatel/Cu2p3/2值分别为0.28、0.27,表明CuO/CeO2催化剂含有更多的Cu+。这是由于,CuO与CeO2间存在金属载体相互作用,形成更多的氧缺陷,有利于Cu+物种的存在,进而提高CO氧化活性。CuO/CeO2-400和CuO/CeO2-600催化剂中Ce3+含量、Vo含量及Cusatel/Cu2p3/2值基本不变,表明焙烧温度不会影响CuO与CeO2间的金属载体相互作用。
2.7 CO氧化性能评价
为了探究载体和催化剂的CO氧化性能,对Al2O3-550载体、CuO/Al2O3催化剂、CeO2-350载体、CuO/CeO2催化剂进行CO氧化活性测试,结果如图7所示。

图7 载体和催化剂的起燃曲线Fig.7 Light-off curves of supports and catalysts
由图7可知,Al2O3-550载体在整个氧化反应过程中并未表现出明显的CO氧化活性。CuO/Al2O3催化剂在180 ℃时开始起活(表现出CO氧化活性),380 ℃时CO基本转化完全,其CO氧化活性较Al2O3-550载体显著提高,表明CuO是CO氧化的活性组分。CuO/Al2O3-600催化剂的起燃曲线T50(CO转化率为50%时的温度)比CuO/Al2O3-400催化剂的高约20 ℃,CuO/Al2O3催化剂的CO氧化活性随焙烧温度的升高略微下降。CeO2-350载体在250 ℃时开始起活,600 ℃时CO基本转化完全,其CO氧化活性较Al2O3-550载体显著提高,这是由于CeO2-350载体表面存在氧缺陷,有利于分子氧在表面活化。CuO/CeO2催化剂在50 ℃时开始起活,起活温度较CeO2-350载体降低约200 ℃,200 ℃时CO基本转化完全,表现出较高的CO氧化活性。CuO/CeO2-600催化剂的起燃曲线T50比CuO/CeO2-400催化剂的仅降低约5 ℃,两者的CO氧化活性基本一致,表明CuO/CeO2催化剂经过高温焙烧能够稳定存在。CuO/CeO2催化剂较CuO/Al2O3催化剂的CO氧化活性显著提高,这是由于,CuO与CeO2间存在金属载体相互作用,能够形成更多的氧缺陷,进而提高CO氧化活性。
3 结论
Al2O3-550载体在低温区间对CO氧化无活性,负载CuO后得到CuO/Al2O3催化剂,其比表面积得到基本保持;CuO在Al2O3-550载体上高度分散,焙烧后无明显烧结,CO氧化活性显著提高。使用CeO2-350载体负载CuO制备得到CuO/CeO2催化剂,高温焙烧下比表面积能够保持,未发生烧结现象;CuO/CeO2催化剂在50 ℃时开始起活,200 ℃时CO基本完全转化,其CO氧化活性较CuO/Al2O3催化剂提高一个数量级以上;600 ℃焙烧的CuO/CeO2催化剂起燃曲线的T50比400 ℃焙烧的降低约5 ℃,具有较好的高温热稳定性。各项表征结果表明,CeO2能够在Ce3+和Ce4+之间转变,与CuO间存在相互作用,容易形成更多的氧缺陷位来稳定Cu+,有利于提高CO氧化活性。
猜你喜欢
发明与创新(2023年30期)2023-10-11 01:37:12
探测与控制学报(2023年4期)2023-09-12 07:26:12
小学生学习指导(高年级)(2023年3期)2023-03-31 06:03:22
华人时刊(2022年9期)2022-09-06 01:02:44
小学生学习指导(高年级)(2022年3期)2022-03-29 07:49:16
分析科学学报(2021年3期)2021-07-14 01:51:16
色谱(2021年6期)2021-05-06 02:18:56
华人时刊(2020年15期)2020-12-14 08:10:36
科技资讯(2020年12期)2020-06-03 04:44:20
小学生导刊(高年级)(2017年2期)2017-06-10 02:40:42
