
QuEChERS-气相色谱-串联质谱法高通量筛查植物源中药材中460种农药残留
2021-08-26 10:51黄学者崔宗岩纪涌彦曹彦忠
农药学学报 2021年4期
黄学者, 崔宗岩, 葛 娜, 纪涌彦, 曹彦忠
(秦皇岛海关技术中心,河北 秦皇岛 066004)
我国中药材种类繁多,常用植物源中药材有600多种,每年有大量的中药材出口到日本、美国等近100个国家和地区[1]。原国家食品药品监督管理总局发布的2013—2016年全国药品质量抽验监测数据表明,国内中药材与饮片总体合格率分别为64%、68%、75%和77%,合格率虽呈逐年提升,但不合格率依然较高,其中农药残留和重金属超标是最主要的风险因素,占80%以上[2]。究其原因,首先,我国中药材生产科技水平不够发达,现有登记用于中药材的农药产品数量有限[3],农民存在滥用、误用农药现象,易导致农药残留风险。其次,目前国内虽然有一些中药材的生产管理规定,但缺乏统一标准,可操作性较差。我国药用和芳香植物生产管理规范 (GAP) 标准还没有推行[4],GAP种植基地较少,规范化种植的中药材仅50多种。因此,亟需建立一种广泛适用于各种类型植物源中药材农药残留的筛查方法。
关于中药材中农药残留检测的前处理方法主要有:固相萃取 (SPE)[5]、在线凝胶渗透色谱(GPC)[6]、基质固相分散 (MSPD)[7]、QuEChERS[8]等,其中QuEChERS方法具有快速、简单、廉价、有效、可靠和安全等特点,得到了越来越多检测机构的认可和广泛应用,食品安全国家标准[9]即采用QuEChERS方法作为农药残留的前处理方式。仪器检测方法主要包括:气相色谱-电子捕获检测法 (GC-ECD)[10]、大气压气相色谱-飞行时间质谱法 (APGC-QTof)[11]、超高效液相色谱-四极杆/轨道阱高分辨质谱联用法 (UPLC-Q-Orbitrap HRMS)[12]、气相色谱-质谱法 (GC-MS)[13]、气相色谱-串联质谱法 (GC-MS/MS)[14-15]、高效液相色谱法(HPLC)[16]、液相色谱-串联质谱法 (LC-MS/MS)[17]等。其中GC-MS/MS具有高抗干扰能力、高通量和高选择性等优点,适用于分析背景干扰严重、定性困难、样品组分含量低的情况,能够满足中药材中农药多残留的检测。
现有中药材农药残留检测方法的不足在于:1) 检测目标物种类有限,高通量多残留检测方法较少;2) 检测样品多集中在一种或一类中药材上,不能满足涵盖各种类型植物源中药材的快速和高通量检测要求。
本研究建立了460种农药残留的GC-MS/MS方法数据库,结合QuEChERS检测方法,分别对5类代表性植物源中药材的提取净化条件进行了考察和优化,并将其应用于市售中药材样品的筛查检测。
1 材料与方法
1.1 仪器与试剂
气相色谱-串联质谱仪 (TSQ 8000型,美国Thermo Fisher公司),高速冷冻离心机 (3-30K型,美国Sigma公司),振荡器 (SA300型,日本Yamato公司),氮吹仪 (N-EVAPTM112型,美国Organomation公司),超纯水发生器 (Mili-Q型,美国Millipore公司),分析天平 (XP105型,瑞士Mettler Toledo公司) 等。
460种农药(见表1)标准品 (纯度≥97%,德国Dr. Ehrenstorfer GmbH、美国Strem Chemicals及美国Sigma Aldrich等公司)。准确称取适量标准品,分别用甲苯等溶剂配制成1 mg/mL的标准储备液,于 −20 ℃保存。标准工作溶液采用甲苯稀释,配制质量浓度为1.0 μg/mL。
甲苯、正己烷和乙腈 (均为色谱纯,Dikma公司);乙酸 (色谱纯,美国TEDIA公司);无水硫酸镁和氯化钠 (分析纯,国药集团化学试剂有限公司);N-丙基乙二胺 (PSA) 和C18(Dikma公司);石墨化碳黑 (GCB,CNW公司)。
1.2 试验方法
1.2.1 样品前处理 准确称取中药材样品2.50 g于50 mL离心管中,加入15 mL去离子水浸泡2 h;加入20.0 mL 0.1%乙酸-乙腈(0.1%为体积分数,下同)、6.00 g无水硫酸镁和2.50 g氯化钠,涡旋混匀2 min,振荡提取20 min,于3 500 r/min下离心5 min;取10.0 mL上清液于40 mL塑料具塞离心管中,管内预先装有1.50 g无水硫酸镁(150 mg/mL)、0.50 g PSA (50 mg/mL)和0.50 g C18(50 mg/mL),涡旋2 min,于10 000 r/min下离心5 min;取5 mL上清液于15 mL玻璃试管中,在45 ℃下氮吹至近干,加入100 μL 7 μg/mL的环氧七氯正己烷溶液,涡旋混匀,过0.22 μm有机相滤膜,待GC-MS/MS检测。
1.2.2 仪器检测条件
色谱条件:DB-5MS型5%苯基-95%甲基聚硅氧烷固定相的弱极性色谱柱 (30 m × 0.25 mm,0.25 μm),进样口温度290 ℃,不分流进样,载气为高纯氦气 (≥99. 999%),采用恒流进样模式,流速1.5 mL/min,进样体积为1.0 μL。升温程序:初始温度为50 ℃,保持1 min,以 20 ℃/min的速率升至100 ℃,然后再以10 ℃/min 的速率升至290 ℃,保持10 min。
质谱条件:EI离子源,温度250 ℃,传输线温度为280 ℃,多离子反应监测 (SRM) 模式。460种农药的保留时间和质谱条件见表1。
2 结果与讨论
2.1 仪器检测方法数据库的建立
2.1.1 色谱条件的优化 选择最常用的DB-5MS型5%苯基-95%甲基聚硅氧烷固定相的弱极性色谱柱,该色谱柱耐高温,适用于不同沸点和极性农药残留化合物的分离和检测。由于串联质谱对目标物的选择性检测能力大大提高,对分离的要求相对降低,因而主要考虑了低沸点和高沸点化合物的特性,以及异构体的分离特征,对升温程序进行了选择和优化,50 ℃保持1 min后对比了3种升温速率:① 20 ℃/min升至290 ℃;② 10 ℃/min升至290 ℃;③ 20 ℃/min升至100 ℃,然后10 ℃/min升至290 ℃。结果发现:升温程序①分析时间短,但部分同分异构体分离效果不好;升温程序②分离效果较好,但分析时间较长。升温程序③既保证了分离效果又节约了分析时间,在1.2.2节色谱条件下实现了460种农药在色谱柱上较好的分离 (图1),通过色谱不能分开的则依据其特征性质谱参数再进行分离。
2.1.2 质谱参数的优化 采用自动化选择反应监测 (AutoSRM) 功能对化合物的特征离子对 (母离子、子离子) 及对应碰撞能量等进行了优化,优化后的质谱参数见表1。实际样品检测采用SRM方式,通过二级选择,使得目标物检测的特异性大大提高,一方面降低了噪音,提高了检测灵敏度,另一方面也避免了复杂基质的干扰,提高了定性定量的准确性。
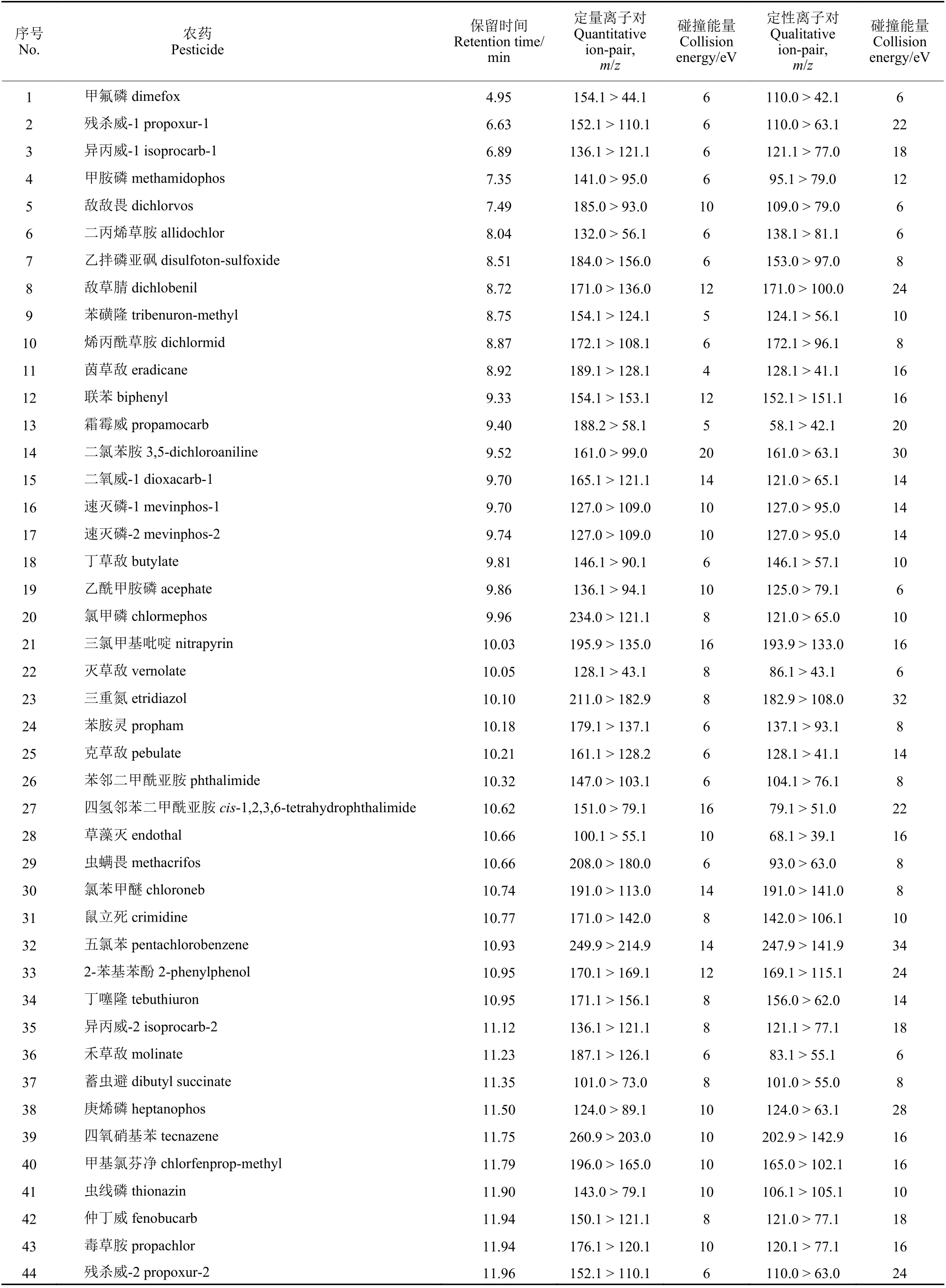
表1 460种农药的保留时间和定量离子对、辅助定性离子对及碰撞能量Table 1 Retention time, quantitative ion, confirming ion and collision energy of 460 pesticides
2.2 QuEChERS方法的选择和优化
针对不同类型的植物源中药材基质,结合国家相关标准[18-22],选择和优化了QuEChERS前处理条件,主要优化了提取溶剂体系和净化条件。
2.2.1 提取条件选择 乙腈是农药残留常用提取试剂,QuEChERS方法提取体系通常分为3种:单一乙腈体系[23]、加乙酸盐缓冲体系[24](美国化学家分析家协会-AOAC标准方法) 以及加柠檬酸盐体系[25]等,其中乙酸盐缓冲体系在实际操作中抗基质效应更强,为了提高对酸碱敏感农药的回收率,本研究选定乙酸-乙腈作为提取溶剂,分别考察了乙腈、0.1%乙酸-乙腈和1%乙酸-乙腈3个提取体系对5种类型植物源中药材的提取效果。结果表明:在100 μg/kg添加水平下,根茎类、花叶类、籽实类、全草类和皮类5种类型中药材基质均表现出相似的变化趋势。以根茎类为例 (图2A),相对纯乙腈,当提取溶剂为0.1%乙酸-乙腈时,均可使得目标物的回收率及相对标准偏差提高,且对大多数样品而言,0.1%和1%乙酸加入量之间无显著差异,因而最终选用0.1%乙酸-乙腈作为提取溶剂。
2.2.2 净化条件选择 参考QuEChERS方法常用净化条件[14,17],本研究选择分散固相萃取法清除样品中的水分和干扰基质,在用无水硫酸镁清除水分的条件下分别考察了PSA、PSA + C18和PSA + C18+GCB 3种净化体系对5种类型中药材基质的净化效果。结果表明:在100 μg/kg添加水平下,根茎类、花叶类、籽实类、全草类和皮类5种类型中药材基质同样均表现出相似的变化趋势,代表性根茎类样品的净化条件对比见图2B。相对于PSA,PSA + C18对基质较为复杂的花叶类、全草类和皮类样品的净化效果明显增强,对基质相对简单的根茎类和籽实类样品的净化效果略有增强;与PSA + C18相比,PSA + C18+ GCB对部分颜色较深的花叶类和全草类样品的净化效果 (祛除颜色)虽有一定程度增强,但GCB对部分农药 (七氯、五氯苯胺、五氯硝基苯和六氯苯等) 吸附作用过强,导致其回收率大大降低,因而本方法未采用PSA + C18+ GCB的净化组合;PSA中加入C18虽然在视觉上对根茎类和籽实类样品净化效果增强不明显,但在不影响回收率的前提下,考虑到尽量减轻连续及大量进样对色谱柱和仪器造成的污染和损害,最终决定选用PSA + C18净化组合。

续表1Table 1 (Continued)

续表1Table 1 (Continued)
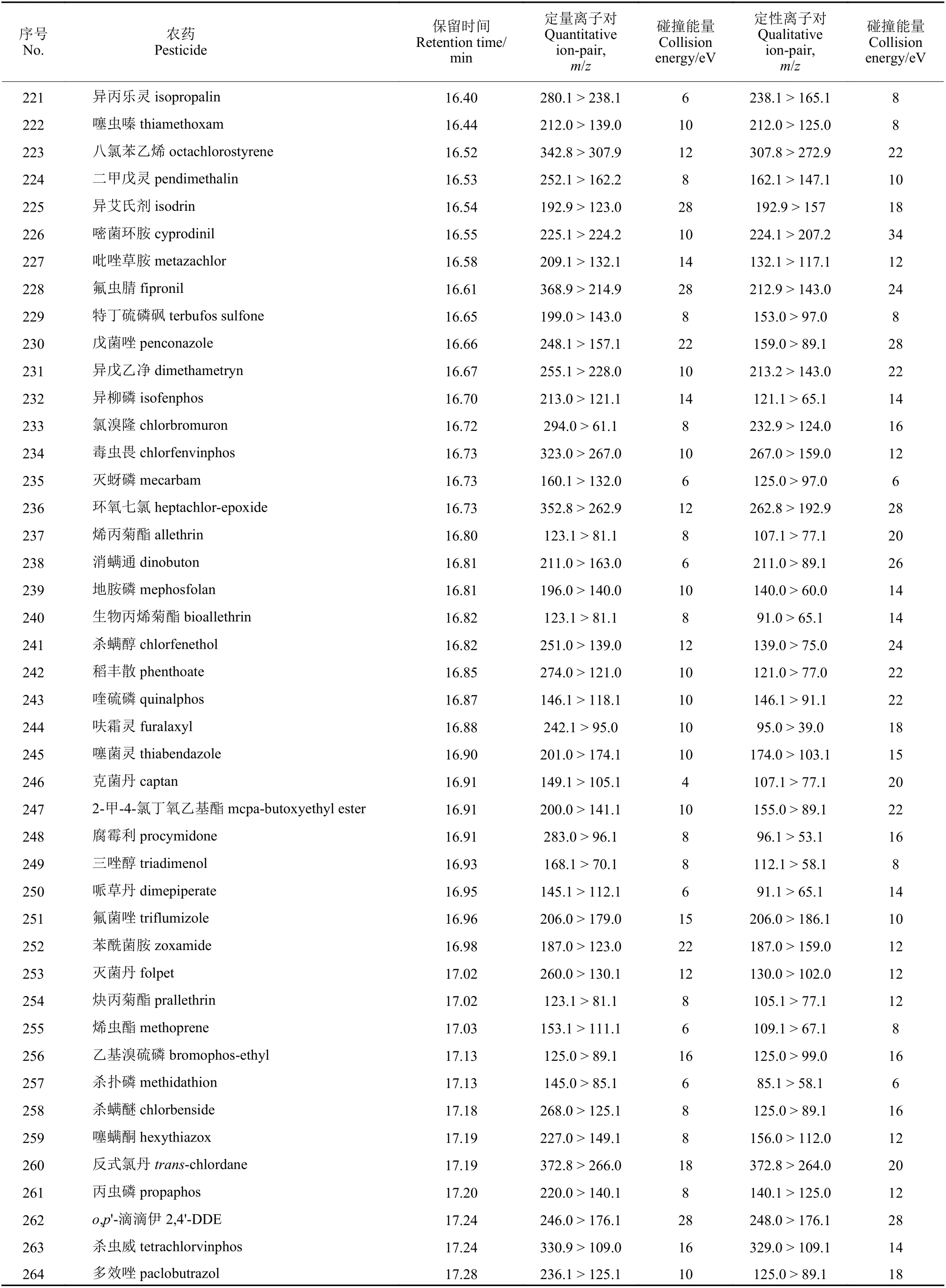
续表1Table 1 (Continued)
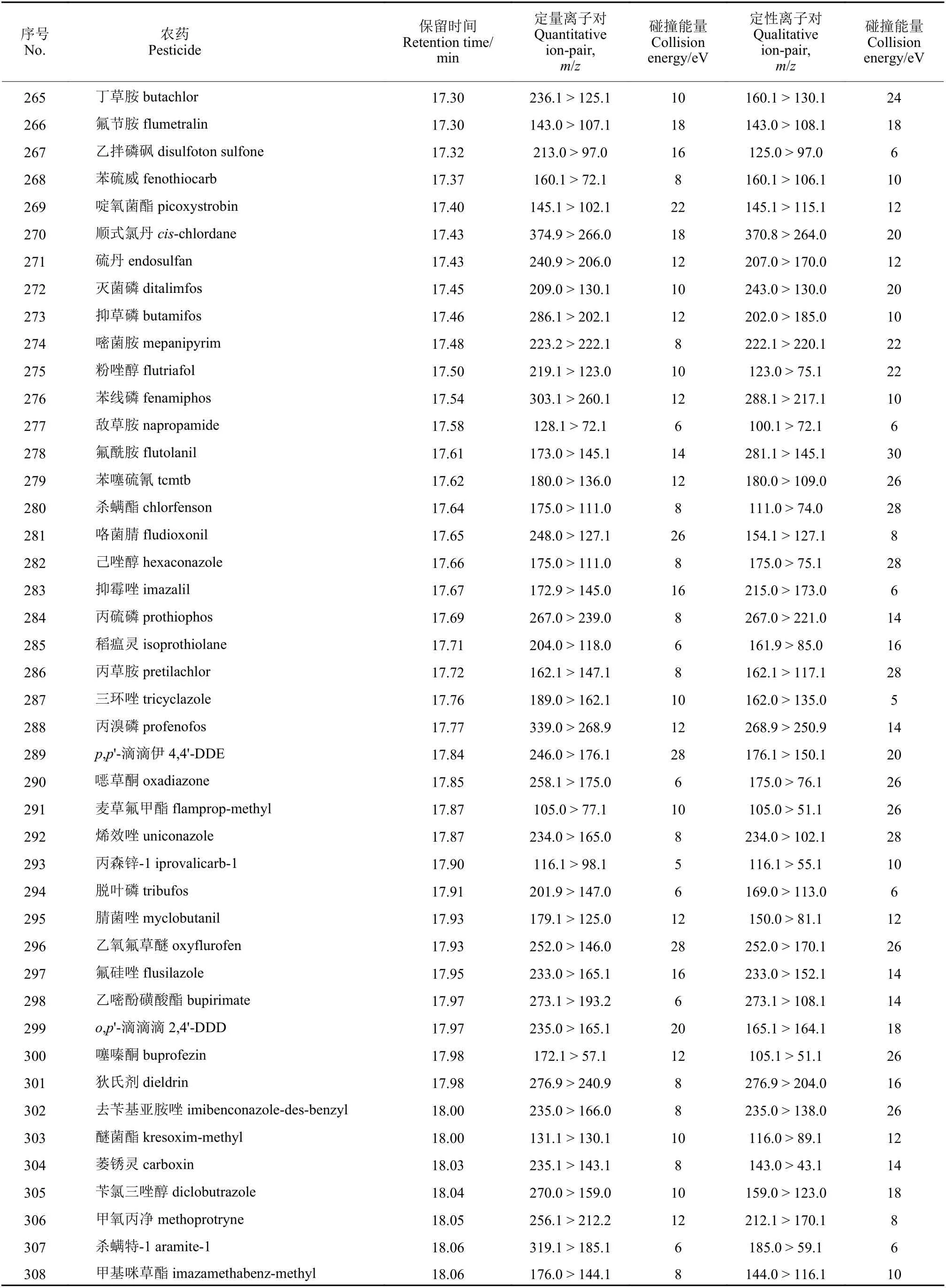
续表1Table 1 (Continued)
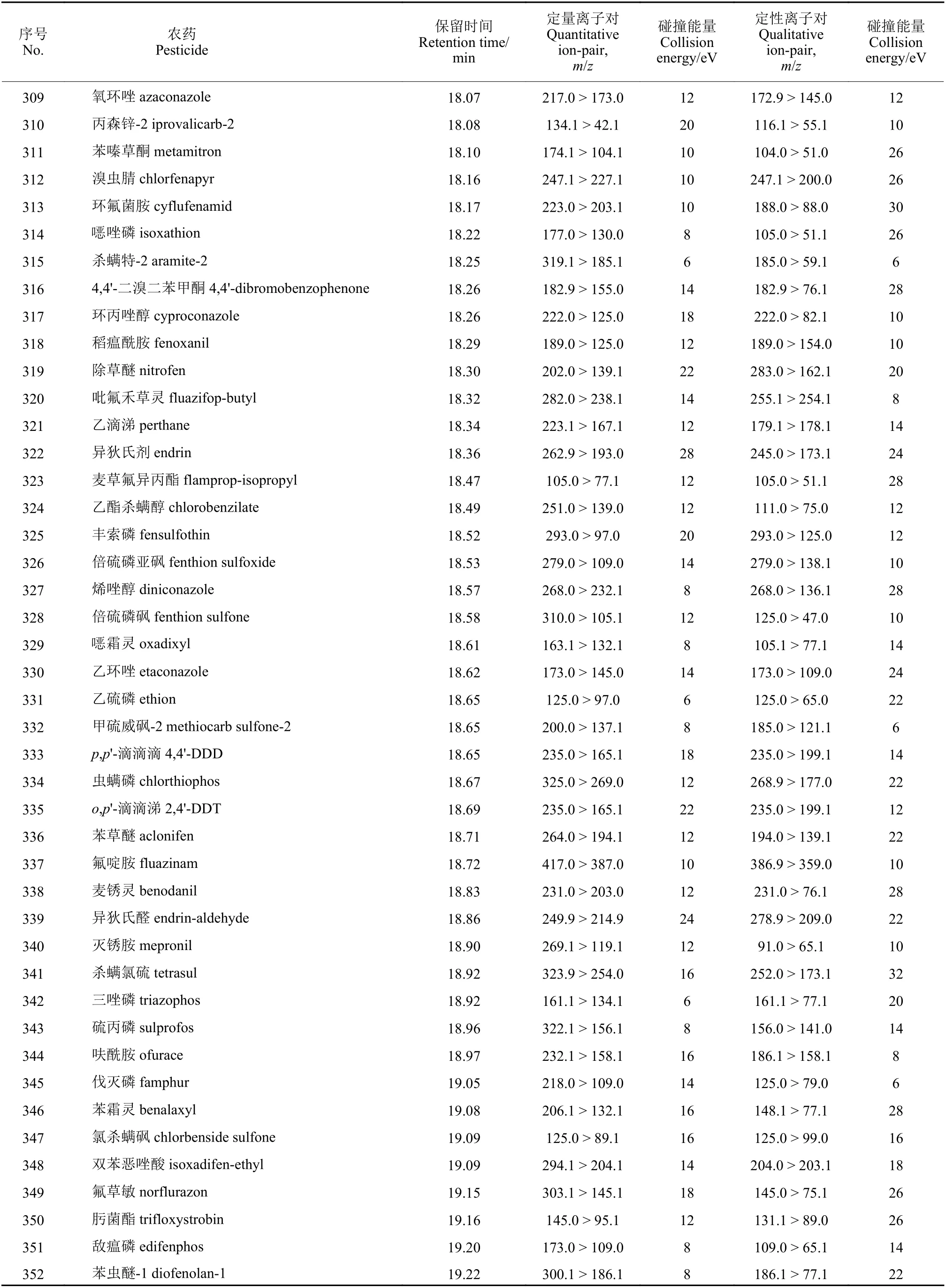
续表1Table 1 (Continued)
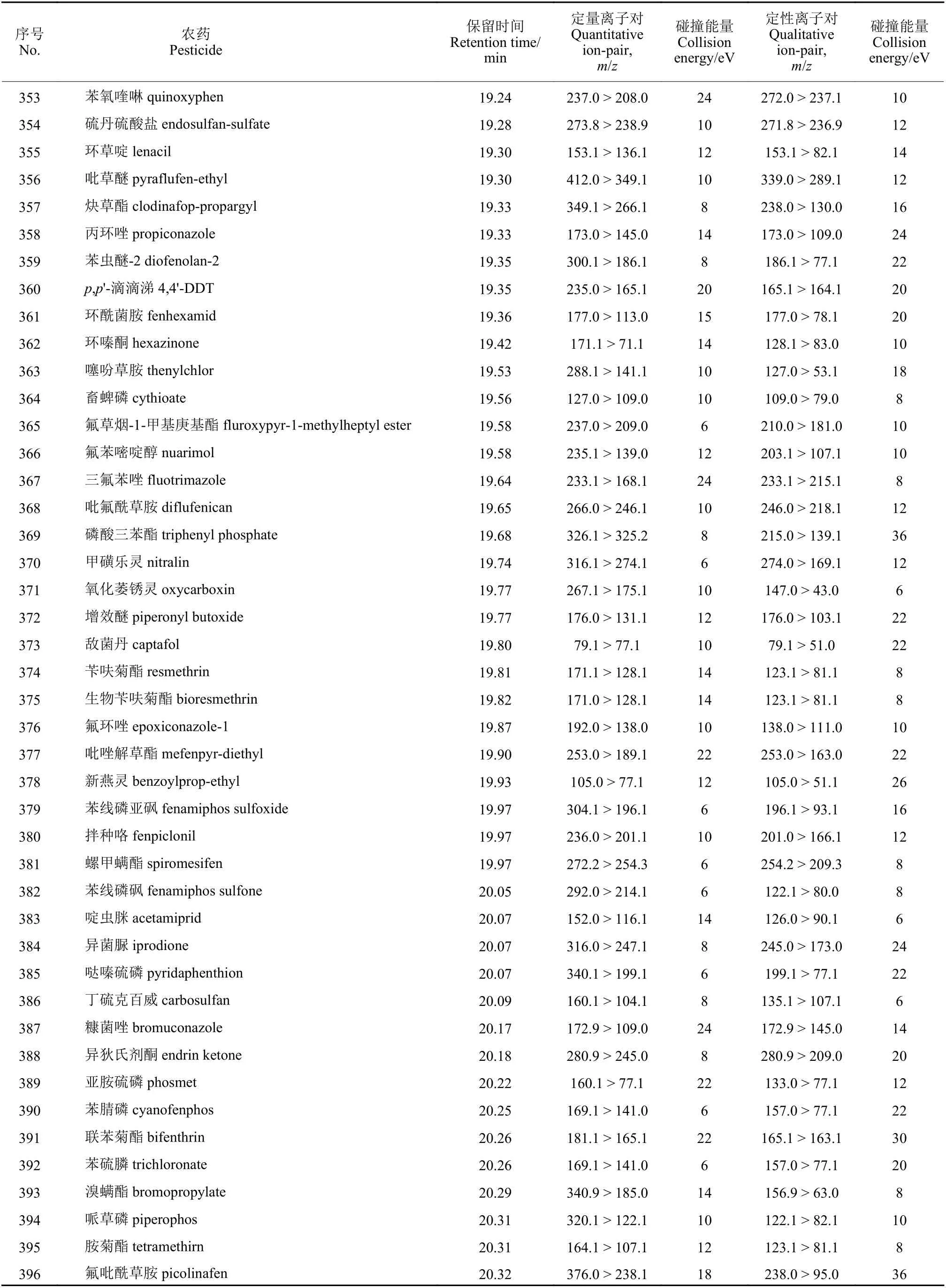
续表1Table 1 (Continued)

续表1Table 1 (Continued)
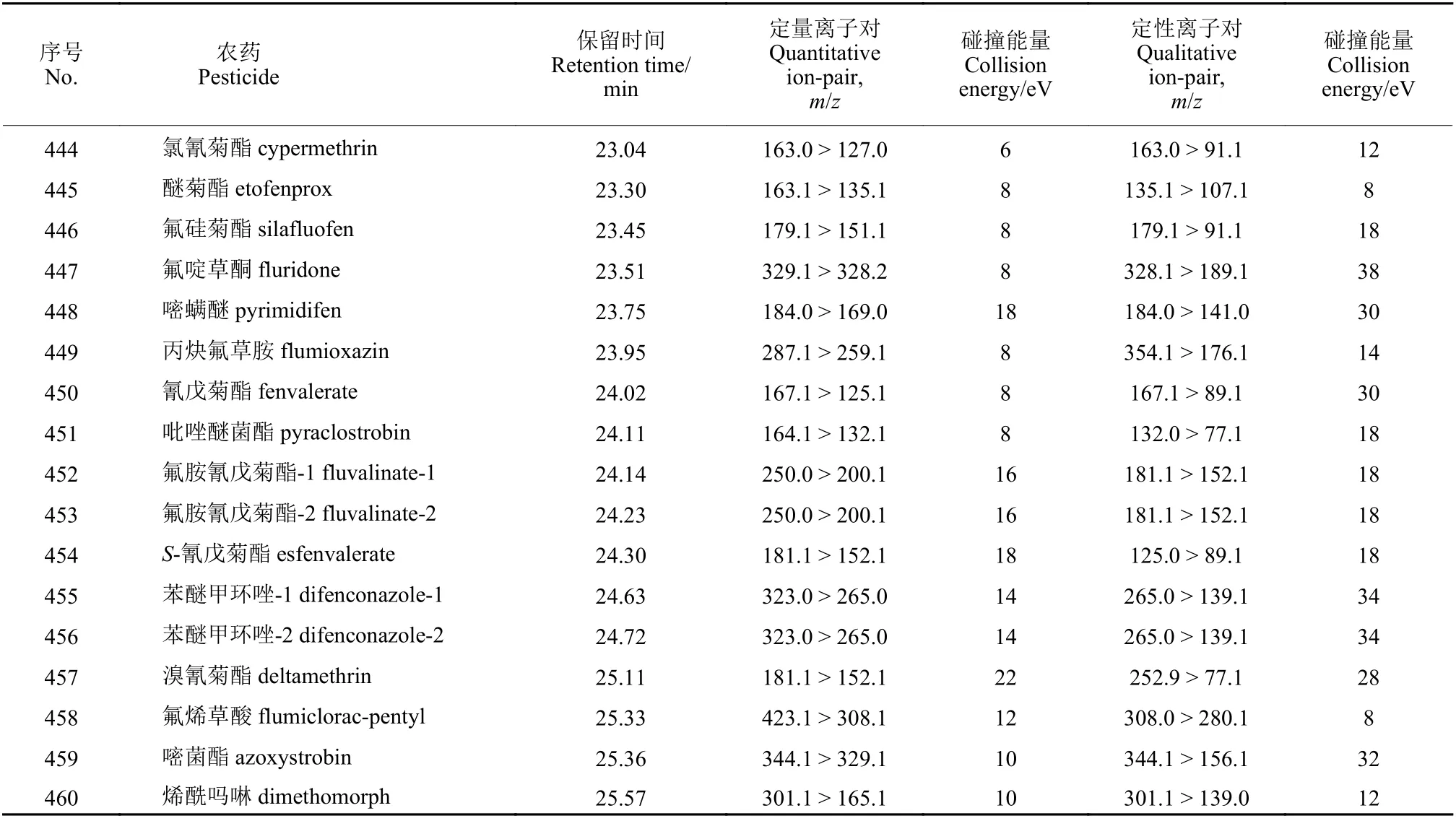
续表1Table 1 (Continued)
2.3 方法性能评价
目前,国内外关于中药材中农药最大残留限量 (MRLs) 的标准主要以药典为准[26]。《欧洲药典》 (EP 8.0)、《美国药典》 (USP 38) 和《英国药典》 (BP 2015) 共涉及中草药中76种农药残留,其中《英国药典》对植物源中药材中的农药进行了概括性限制:有机氯类农药的MRL值为0.05 mg/kg,其他农药MRL值为0.5 mg/kg 或1.0 mg/kg;《中国药典》规定了22种有机氯类农药的MRL值。本研究考察了QuEChERS-GC-MS/MS方法在5种类型中药材基质中的定量限 (LOQ) (信噪比为10时对应的目标物浓度),表2中仅列出了其中至少在一种基质中LOQ大于50 μg/kg的农药品种。

表2 QuEChERS-GC-MS/MS测定5种类型中药材基质中农药残留的定量限 (仅列出LOQ大于50 μg/kg的农药)Table 2 Limits of quantification (LOQ) for determination of pesticide residues in different types of Chinese medicinal materials by QuEChERS-GC-MS/MS (Only pesticides of LOQ > 50 μg/kg were listed) (μg/kg)
总体而言,对不同类型中药材样品,本方法的LOQ整体上在0.006~221 μg/kg之间,其中80%以上目标物LOQ不高于50 μg/kg,对于根茎类和籽实类样品,60%以上农药LOQ低于10 μg/kg,说明本筛查检测方法在高通量基础上具有较高的灵敏度;针对不同类型植物源中药材样品所测得的回收率和相对标准偏差数据表明,72%以上目标物的回收率在60%~120%之间,77%以上农药的相对标准偏差在20%以内,方法的准确度良好。
与现有国家标准GB 23200.10—2016[18]及其他相关文献方法相比,本研究建立的QuEChERSGC-MS/MS方法更加全面适用于不同类型植物源中药材基质 (根茎类、花叶类、籽实类、全草类、皮类等),可实现一次进样同时检测400种以上农药残留,且大多数农药的灵敏度和准确度均可满足方法学要求[27],具有简便、快速和高通量的优点,适用于不同类型植物源中药材中农药残留的筛查测定。
2.4 实际样品检测
为了验证本研究方法的实用性并系统了解市售植物源中药材中农药残留状况,采用本研究方法对从药店及药材市场等地采集的215批中药材样品进行了筛查检测。结果表明,有17批样品未检出任何农药残留,其余198批样品均检出1种以上农药残留,占检测样品总数的92%。其中所有花叶和全草类样品均检出了农药残留,可能与这些类型的中药材为农药直接施用部位有关。从检出农药种类来看,所有样品共检出72种农药残留,检出率最高的3种农药分别为毒死蜱 (20.7%)、氟氯氰菊酯 (16.0%) 和六氯苯 (15.3%),其中毒死蜱最高残留量达到355 μg/kg,氟氯氰菊酯最高残留量达到1 014 μg/kg,应当引起关注和重视,可以作为其质量安全评价重点关注的风险物质。
3 结论
建立了植物源中药材中农药多残留高通量筛查检测的QuEChERS-GC-MS/MS。样品在用0.1%乙酸-乙腈提取、氯化钠盐析分层、PSA + C18分散固相萃取净化条件下,针对根茎类、花叶类、籽实类、全草类和皮类5种类型中药材基质,实现了一次进样同时检测400多种农药残留。该方法具有快速、灵敏和高通量的优势,将其运用于市售215批植物源中药材样品检测,发现毒死蜱、氟氯氰菊酯等农药检出率较高,应当重点关注和防控。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国高原医学与生物学杂志(2022年3期)2022-06-22
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
今日农业(2021年12期)2021-11-28
今日农业(2021年7期)2021-11-27
今日农业(2021年21期)2021-11-26
国际太空(2021年4期)2021-06-18
土木与环境工程学报(2021年1期)2021-03-06
国际太空(2019年12期)2019-02-07
中国信息化·学术版(2013年3期)2013-06-25
