
酰胺修饰杂化硅胶整体柱的制备及其在莱克多巴胺检测中的应用
2021-08-24 13:31彭传云张少文吴春来赵晓洁宋根娣
分析测试学报 2021年8期
彭传云,张少文,冯 勇,吴春来,赵晓洁,宋根娣
(洛阳理工学院 环境工程与化学学院,河南 洛阳471023)
莱克多巴胺(RAC)属于人工合成的苯乙醇胺类药物,是一种“瘦肉精”并作为克伦特罗的替代品被非法添加在饲料中,用来促进动物(如猪、牛、羊等)体内蛋白质含量的提高,使脂肪分解代谢增强,提高瘦肉率[1]。莱克多巴胺在鲜肉及肉制品中残留,会对人体产生一定毒副作用。我国已明确禁止在动物养殖过程中使用“瘦肉精”,但违法滥用现象时有发生,因此建立有效的检测方法对于控制食品安全具有重要意义。
目前检测莱克多巴胺的方法主要有高效液相色谱法(HPLC)[2]、电化学分析法[3]、高效液相色谱-串联质谱法(HPLC-MS)[4-5]和酶联免疫法(ELISA)[6-7]等。其中,HPLC法由于具有灵敏、快速、高效和操作方便等优点被广泛应用于莱克多巴胺的检测。由于肉样基质成分复杂,影响分析检测的灵敏度和准确度,在实际样品检测中减小基质效应可有效提高检测的准确度。基质添加标准曲线法和优化样品前处理条件均可有效消除基质效应的影响[8-10]。基质添加标准曲线法是以空白样品为基质建立标准曲线,降低基质效应[11]。采用固相微萃取技术对实际样品前处理也是去除基质中杂质的常见方法[12-14],例如,以聚(甲基丙烯酸-乙二醇二甲基丙烯酸酯)整体柱为固相微萃取介质,对猪肉样品中的β-受体激动剂进行富集,可有效去除猪肉中的基质干扰,对莱克多巴胺的检出限低至0.5 μg/kg[15]。近年来,有机-无机杂化材料因其较高的萃取效率、机械稳定性和易被修饰改性的特性,常用作实际样品前处理材料[16-19]。分子印迹聚合物复合Fe3O4材料和Fe3O4印迹金属有机框架MOF-5材料均被用于不同实际样品(如猪肉、猪肝、尿液、饲料和毛发)的前处理,实现莱克多巴胺的快速准确检测[20-21]。
本文利用溶胶-凝胶法制备酰胺修饰氧化琼脂糖AG/SiO2整体柱,并以此为固相微萃取介质,应用于肉样中莱克多巴胺的萃取富集,通过优化实际样品前处理条件,结合高效液相色谱-紫外检测,建立了一种灵敏、可靠的实际样品中莱克多巴胺的分析方法。
1 实验部分
1.1 仪器与试剂
岛津LC-20AT液相色谱仪(日本岛津公司);3-18K台式高速低温离心机(德国CHRIST冻干机有限公司);DY89-1型电动玻璃匀浆机(宁波新芝生物科技有限公司);XW-80A旋涡混合器(上海青浦沪西仪器厂);LSP04-1A注射泵(河北保定兰格恒流有限公司);Bruker vector 22傅里叶变换红外光谱仪(FTIR,德国Bruker公司);Philips XL30E扫描电子显微镜(SEM,荷兰Philips公司);Escalab 250xi X射线光电子能谱(XPS,赛默飞世尔科技有限公司);ASAP 2020 N2吸附-脱附仪(美国Micromeritics Instruments公司)。
醋酸铵、氨水、磷酸二氢钾、N,N-二甲基甲酰胺(DMF)、盐酸、硝酸(分析纯,天津市科盟化工工贸有限公司);过硫酸钾(K2S2O8)、巯基乙酸(CH3COSH)、琼脂糖(AG)、二环己基碳二亚胺(DCC)、烯丙基缩水甘油醚(AGE)、N-羟基琥珀酰亚胺(NHS)(分析纯,百灵威科技有限公司);甲醇、乙腈(色谱纯,昌泰兴业有限公司);冰乙酸、乙酸乙酯、三氯甲烷、四氯化碳、石油醚、丙酮(分析纯,天津市天力化学试剂有限公司);正己烷(分析纯,天津市大茂化学试剂厂);聚乙二醇(PEG,Mw=10 000)、尿素(分析纯,国药集团化学试剂有限公司);四甲氧基硅烷(TMOS,98%,武汉大学有机硅新材料有限公司);莱克多巴胺标准品(美国Sigma-Aldrich公司);熔融石英毛细管(530 μm I.D.,河北永年锐沣色谱器件公司);实验用水为超纯水;羊肉随机购自附近超市、农贸市场。
1.2 标准溶液的配制
准确称取莱克多巴胺标准品(0.010 0±0.000 5)g,以甲醇溶解并定容至100 mL,即得质量浓度为100 μg/mL的莱克多巴胺储备液,于4℃下保存备用。使用时根据需要以相应的空白样品基质提取溶液配制成适当浓度的标准工作溶液。
1.3 整体柱的制备
毛细管预处理[22]:用1 mol/L NaOH活化毛细管柱,然后分别用0.1 mol/L HCl、水冲洗至中性,氮气吹干备用。
AG氧化:取1.0 g AG与12 mL 3 mol/L HNO3在60℃水浴加热下反应6~7 h,即得氧化AG溶液。
AG/SiO2整体柱制备(见图1A):采用溶胶-凝胶法制备AG/SiO2整体柱。称取0.25 g尿素和0.17 g聚乙二醇溶于3 mL氧化AG溶液,在冰水浴(0℃)下磁力搅拌2 min,用浓氨水调节pH至中性,然后在0℃下搅拌2 h,加入1 mL TMOS搅拌30 min后超声至形成均一溶胶。将其灌装至处理好的石英毛细管中,用硅胶垫将毛细管两端封口,40℃下反应20 h,升温至90℃,保持7~8 h后,降至室温取出。分别用甲醇、丙酮洗去未反应的化合物,干燥后备用。
酰胺基团功能化修饰过程[23](见图1B):首先取1.0 mL AGE注入制备好的AG/SiO2整体柱中,两端封口,40℃下反应16 h,利用开环反应在多糖结构中引入碳碳双键;再将2 mL 0.025 g/mL的K2S2O8和0.000 16 g/mL CH3COSH的混合水溶液注入上述整体柱中,两端封口,60℃反应24 h,通过巯烯点击反应进一步引入羧基;最后取1 mL DCC(0.05 g/mL)和NHS(0.014 g/mL)的DMF混合溶液注入上述修饰后的整体柱,两端封口后于40℃下反应20 h,利用NHS与羧基酰胺化反应对整体柱完成修饰,制得酰胺修饰AG/SiO2整体柱。分别用丙酮和去离子水通入整体柱后,干燥备用。

图1 酰胺修饰AG/SiO2整体柱的制备过程Fig.1 The preparation procedure of amide group functioned AG/SiO2 monolithic column
1.4 实际样品制备
取适量羊肉充分绞碎后匀浆,准确称取2.0 g于25 mL离心管中,加入15 mL乙腈-水(7∶3,体积比)和1 mL乙酸-乙酸铵缓冲溶液(pH 5.6),涡流振荡10 min,超声15 min后,于4℃以8 000 r/min离心5 min。取上清液于25 mL离心管中,加入15 mL四氯化碳,静置30 min后取上清液氮气吹干,残留物用2 mL乙腈充分溶解,待上柱净化。
1.5 固相微萃取过程
以酰胺修饰AG/SiO2整体柱为固相微萃取介质,截取5 cm粘至注射器针头,用注射泵作为驱动,将溶液推入整体柱中,先用1.0 mL甲醇活化20 min(流速为0.05 mL/min),然后取1.0 mL实际样品预处理后的上清液通过整体柱萃取20 min(流速为0.05 mL/min),通空气20 min,再用甲醇-乙酸(9∶1,体积比)洗脱富集在整体柱上的莱克多巴胺0.83 min(流速为0.03 mL/min),取25 μL洗脱液,氮气吹干后溶解至50 μL水中,用于液相色谱分析。
1.6 液相色谱条件
色谱柱:Agilent Zorbax SB-C18(4.6 mm×250 mm,4.6 μm);紫外检测器检测波长:276 nm;流动相:0.005 mol/L磷酸二氢钾水溶液-乙腈(77∶23,体积比),等度洗脱;流速:1.0 mL/min;进样量:20 μL;柱温:25℃;采集时间:10 min。
2 结果与讨论
2.1 整体柱材料的表征
图2 A为酰胺修饰AG/SiO2整体柱XPS碳元素的1 s电子层能谱(C1s能谱)分析。图中C―C(284.7 eV)、C―O(285.7 eV)、O====C(286.8 eV)和O====C―N(290.2 eV)4个峰表明经过功能化修饰反应,酰胺基团被修饰至琼脂糖上。由红外谱图(图2B)可见,经过酰胺基团功能化修饰,在3 332~3 454 cm-1处的吸收峰对应修饰后引入的N―H伸缩振动;2 938 cm-1和2 850 cm-1处的吸收峰分别对应C―H键的伸缩振动;1 247 cm-1和1 576 cm-1处的吸收峰对应C―O―C和C―N特征吸收峰,结合1 629 cm-1处C====O伸缩振动的吸收峰,表明AG/SiO2整体柱修饰了酰胺基团。氮气吸附-脱附等温曲线如图2C所示,运用BET吸附模型(Brunauer-Emmett-Teller)计算得到整体柱材料的比表面积为131.7 m2/g,孔径主要分布在1~2 nm之间。由扫描电镜照片(图2D)可见,整体柱材料与毛细管壁键合良好,且材料有孔,具有良好的通透性。修饰上酰胺基团[24]和较大的比表面积均有利于莱克多巴胺的富集。
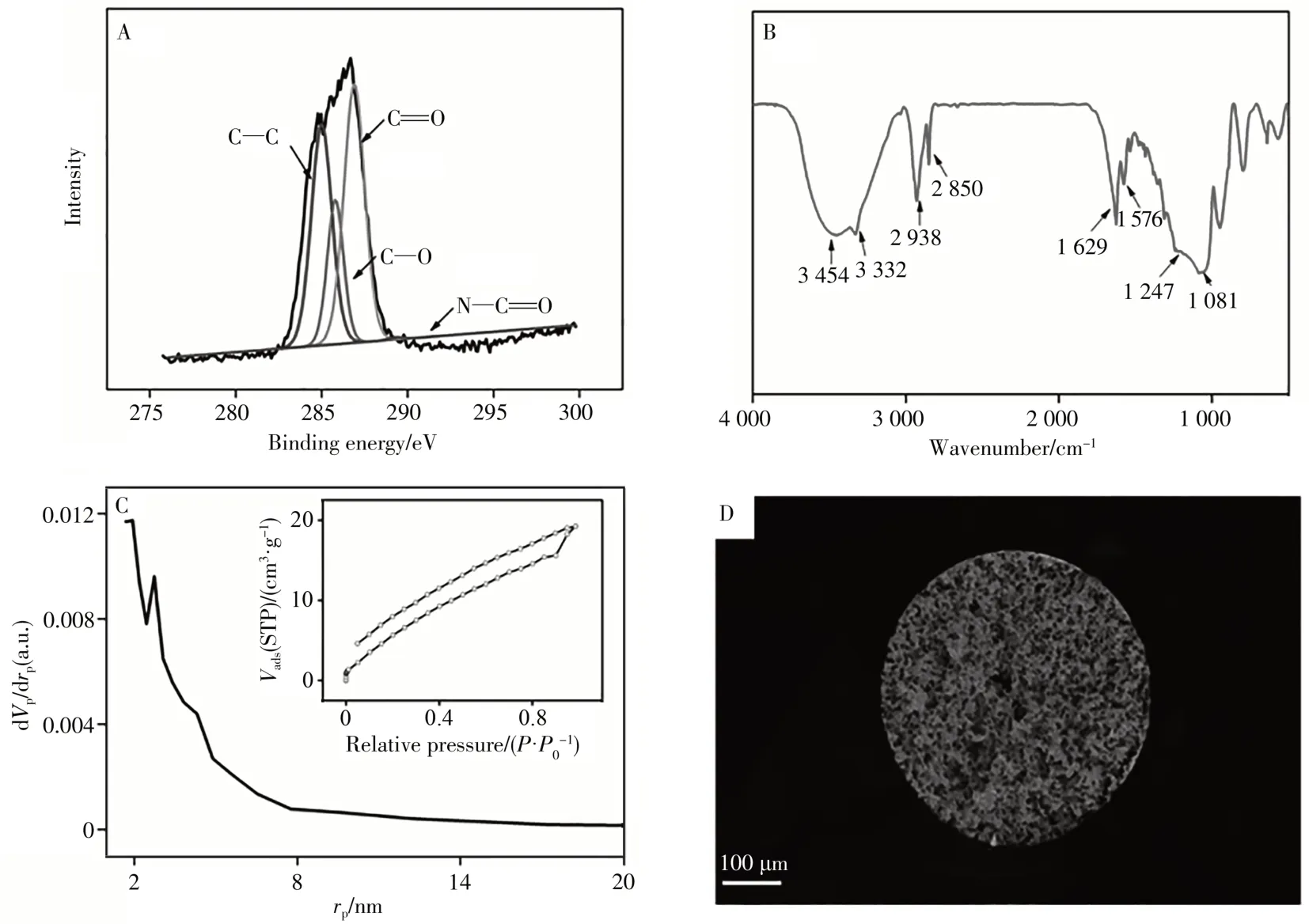
图2 酰胺修饰AG/SiO2整体柱的XPS谱图(A)、红外谱图(B)、N2吸附-脱附曲线和孔径分布图(C),以及SEM扫描电镜照片(D)Fig.2 XPS spectrum(A),FTIR spectrum(B),N2 adsorption-desorption curve and pore size distribution(C)and SEM image(D)of amide modified AG/SiO2 monolithic column
2.2 前处理条件的优化
2.2.1 提取溶剂的优化研究表明,选用乙腈为提取溶剂不但可有效沉淀蛋白质,而且对β-受体激动剂有良好的提取效果[25],同时莱克多巴胺可溶于水,因此实验分别以乙腈、乙腈-水(9∶1)、乙腈-水(8∶2)、乙腈-水(7∶3)、乙腈-水(6∶4)为提取溶剂,考察了其对添加相同浓度莱克多巴胺肉样的提取效果。结果显示,以乙腈-水(7∶3)为提取溶剂时,莱克多巴胺的色谱峰面积最大,提取效率最高,因此选择乙腈-水(7∶3)作为提取溶剂。
2.2.2 pH值的优化样品溶液pH值对莱克多巴胺的存在状态和稳定性有一定影响,进而影响整体柱材料与莱克多巴胺的相互作用。实验考察了pH值分别为4.0、5.2、5.6、6.0、7.0、9.0、10.0和12.0时整体柱对莱克多巴胺萃取效率的影响。结果表明,在pH 5.6时,莱克多巴胺的色谱峰面积最大,提取效率最高,因此实验选择在pH 5.6条件下处理肉样。
2.2.3 萃取与解吸条件的优化考察了上样速率(0.015~0.2 mL/min)对莱克多巴胺萃取效果的影响,结果显示,萃取后莱克多巴胺的色谱峰面积随上样速率的增大而减小,综合考虑萃取效率和萃取时间,选择上样速率为0.05 mL/min。解吸速率也是影响萃取效率的一个重要因素,实验研究了解吸速率(0.005~0.07 mL/min)对莱克多巴胺提取效率的影响,发现解吸速率从0.005 mL/min增至0.03 mL/min时莱克多巴胺的色谱峰面积几乎保持不变,但解吸速率继续增加时,莱克多巴胺的色谱峰面积减小,因此选择最佳解吸速率为0.03 mL/min。在保持其他条件一致的情况下考察了解吸体积(12.5~50 μL)的影响,结果显示,当解吸体积从12.5 μL增至25 μL时莱克多巴胺的色谱峰面积也随之增加,但当解吸体积大于25 μL时,其色谱峰面积几乎不变。表明当解吸体积为25 μL时,富集的莱克多巴胺被完全洗脱,因此选择最佳解吸体积为25 μL。
2.3 整体柱制备的重现性
以50 ng/mL标准溶液为样品,在相同的检测条件下,测定了同批次及不同批次制备的AG/SiO2整体柱对莱克多巴胺的萃取结果,并以相对标准偏差(RSD)评价整体柱制备的重现性。同一批次整体柱检测的RSD为6.6%,不同批次整体柱检测的RSD为7.9%,表明整体柱制备的重现性较好。
2.4 分析方法评价
2.4.1 线性范围及检出限以空白羊肉为基质,将7个不同浓度的莱克多巴胺标准溶液添加至空白羊肉中(添加量为1.94~1 170 ng/g)。在优化的萃取条件下,经整体柱萃取净化后用高效液相色谱分析,并以标准溶液添加量为横坐标(x,ng/g),萃取后莱克多巴胺的色谱峰面积为纵坐标(y),每个样品分别测定3次,绘制标准曲线。结果显示,莱克多巴胺的线性范围为1.94~1 170 ng/g,回归方程为y=5 929.9x+590.04(r2=0.993 1)。分别以3倍和10倍信噪比计算得到方法的检出限(LOD)和定量下限(LOQ),LOD和LOQ分别为0.618、1.94 ng/g。
表1 列举了近年来莱克多巴胺的检测方法与本方法的比较。结果显示,本方法对莱克多巴胺的萃取效果较好,检出限较低,方法灵敏准确。

表1 本方法与其他方法的对比Table 1 Comparison of the established method with other methods
2.4.2 方法准确度与精密度以羊肉为检测对象,通过标准添加实验,以加标回收率和RSD分别考察方法的准确度和精密度。莱克多巴胺的加标水平为0.59、1.18、2.35 ng/g,每个加标水平做5个平行实验。结果表明,3个加标水平下,莱克多巴胺的平均回收率为86.6%~103%,RSD为4.8%~7.3%,方法的准确度和精密度满足微量检测需求。加标羊肉样品的色谱图见图3。
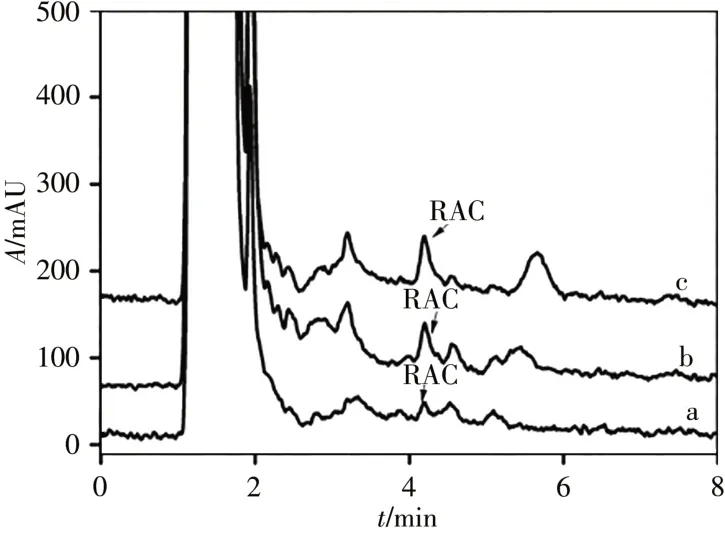
图3 加标0.59 ng/g(a)、1.18 ng/g(b)和2.35 ng/g(c)的羊肉样品经萃取后的色谱图Fig.3 Chromatograms of the mutton samples spiked at 0.59 ng/g(a),1.18 ng/g(b)and 2.35 ng/g(c)ractopamine
2.5 实际样品检测
采用本文建立的方法对36批次羊肉样品进行检测,每个样品平行测定3次。结果表明,有2批次羊肉中检出莱克多巴胺,含量分别为0.17、2.51 μg/g,其余样品中均未检出。
3 结论
本文以酰胺修饰AG/SiO2整体柱为固相微萃取介质,优化了样品前处理条件,建立了肉样中莱克多巴胺的HPLC-UV检测方法。结果表明,实际样品经乙腈-水(7∶3)提取及乙酸-乙酸铵缓冲溶液(pH 5.6)处理,提取液经整体柱萃取后,可有效去除样品中的复杂基质,实现对莱克多巴胺的净化和富集。所建立的检测方法在1.94~1 170 ng/g范围内呈良好的线性关系,r2为0.993 1,LOD为0.618 ng/g,LOQ为1.94 ng/g。加标回收率为86.6%~103%,RSD为4.8%~7.3%。该方法具有检出限低、回收率高、样品用量少等特点,并成功应用于羊肉中莱克多巴胺的检测,可为肉样中痕量“瘦肉精”的检测提供技术支持。
猜你喜欢
中国造纸(2022年8期)2022-11-24
流程工业(2022年3期)2022-06-23
流程工业(2022年5期)2022-06-23
商业评论(2022年4期)2022-05-05
读者(2022年9期)2022-04-22
老年医学研究(2021年5期)2022-01-19
疯狂英语·新读写(2020年3期)2020-06-06
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
时代英语·高一(2019年5期)2019-09-03
