
Argon reduces microglial activation and inflammatory cytokine expression in retinal ischemia/reperfusion injury
2021-08-20 12:37:20UlrichGoebelStefanieScheidSashkoSpassovNilsSchallnerJakobWollbornHartmutBuerkleFelixUlbrich
中国神经再生研究(英文版) 2021年1期
Ulrich Goebel, Stefanie Scheid, Sashko Spassov, Nils Schallner, Jakob Wollborn,Hartmut Buerkle, Felix Ulbrich
Abstract We previously found that argon exerts its neuroprotective effect in part by inhibition of the toll-like receptors (TLR) 2 and 4. The downstream transcription factors signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa B (NF-κB) are also affected by argon and may play a role in neuroprotection. It also has been demonstrated that argon treatment could mitigate brain damage, reduce excessive microglial activation, and subsequently attenuate brain inflammation. Despite intensive research, the further exact mechanism remains unclear. In this study, human neuroblastoma cells were damaged in vitro with rotenone over a period of 4 hours (to mimic cerebral ischemia and reperfusion damage), followed by a 2-hour post-conditioning with argon (75%). In a separate in vivo experiment, retinal ischemia/reperfusion injury was induced in rats by increasing intraocular pressure for 1 hour. Upon reperfusion, argon was administered by inhalation for 2 hours. Argon reduced the binding of the transcription factors signal transducer and activator of transcription 3, nuclear factor kappa B, activator protein 1, and nuclear factor erythroid 2-related factor 2, which are involved in regulation of neuronal damage. Flow cytometry analysis showed that argon downregulated the Fas ligand. Some transcription factors were regulated by toll-like receptors; therefore, their effects could be eliminated,at least in part, by the TLR2 and TLR4 inhibitor oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (OxPAPC). Argon treatment reduced microglial activation after retinal ischemia/reperfusion injury. Subsequent quantitative polymerase chain reaction analysis revealed a reduction in the pro-inflammatory cytokines interleukin (IL-1α), IL-1β, IL-6, tumor necrosis factor α, and inducible nitric oxide synthase.Our results suggest that argon reduced the extent of inflammation in retinal neurons after ischemia/reperfusion injury by suppression of transcription factors crucial for microglial activation. Argon has no known side effects or narcotic properties; therefore, therapeutic use of this noble gas appears ideal for treatment of patients with neuronal damage in retinal ischemia/reperfusion injury. The animal experiments were approved by the Commission for Animal Care of the University of Freiburg (approval No. 35-9185.81/G14-122) on October 19, 2012.
Key Words: argon; ischemia/reperfusion injury; microglia; neuroinflammation;neuroprotection; noble gas; SH-SY5Y; toll-like receptor; transcription factor
Introduction
Damage due to cerebral ischemia and reperfusion damage can destroy nerve tissues, leading to loss of function and increased mortality in affected patients. Anesthesia or surgery can reduce the blood supply, with resulting severe impairments that can manifest as delirium, cognitive dysfunction, or even stroke (Wesselink et al., 2018). At the cellular level,inflammatory and apoptotic processes play a major role in the deterioration of neurons (Ahmad et al., 2014). Neuronal cells have only a limited potential for regeneration, making cerebral ischemia and reperfusion injuries a major cause of morbidity and mortality (Ng et al., 2011). At present, no measures are available to counteract these degenerative processes.
In recent decades, a large number of drugs have been investigated for their potential neuroprotective effects against cerebral ischemia/reperfusion injury, predominantly in preclinical analyses, but most have had disappointing results.One exception is the noble gas argon, which has shown mostly positive results in terms of neuroprotection (Ulbrich et al.,2014, 2015a; Moretti et al., 2015). The protective effect of argon on neuronal tissue has been demonstrated in variousex vivoandin vivostudies (Soldatov et al., 1998; Loetscher et al., 2009; Ryang et al., 2011; David et al., 2012; Fahlenkamp et al., 2012; Brucken et al., 2014; Ulbrich et al., 2014, 2015a).However, the molecular mechanism of action of argon has not been established.
We have demonstrated that argon has anin vivoanti-apoptotic effect (Ulbrich et al., 2014, 2015a, 2016). After ischemia/reperfusion injury of the retina, argon significantly reduced the death of retinal ganglion cells (Ulbrich et al., 2014). In addition, argon treatment activated extracellular regulated kinase 1/2 (ERK1/2), resulting in decreased expression of heme oxygenase-1 (Ulbrich et al., 2015a). Zhuang et al. (2012)also showed an effect of argon on the anti-apoptotic signaling pathway in a neonatal asphyxia model of the rat in the form of increases in B-cell lymphoma-2/xL (Bcl-2/Bcl-xL) levels and decreases in Bcl-2-associated x protein levels in brain tissue(Zhuang et al., 2012). Fahlenkamp et al. (2012) conducted anin vitroinvestigation of the molecular mechanism by examining the influence of argon on primary neuronal and astroglial cell cultures, as well as on the microglial cell line BV-2. They found that argon treatment increased the phosphorylation of ERK1/2, and they reported, for the firsttime, an anti-inflammatory effect of argon via modulation of the expression of the pro-inflammatory cytokine IL-1β.
In a previous study, we exposed human neuroblastoma cells to argon following damage by rotenone (Ulbrich et al.,2015b). We found that argon exerts its neuroprotective effect in part by inhibition of the toll-like receptors (TLR2) and TLR4.The downstream transcription factors signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa B (NF-κB) are also affected by argon and may play a role in neuroprotection (Przanowski et al., 2014; Ulbrich et al.,2014; Mouihate and Mehdawi, 2016). In cerebral injuries, the neuroprotective effect of argon appears to involve microglia,which are the resident immune cells of the central nervous system. Changes in microglial morphology and function are related to signaling molecules that are released in response to injury stimuli, such as stroke. For example, microglia aid in the elimination of dead neuronal cells and in the restoration of the delicate milieu within the central nervous system. However,excessive activation of microglia by damage-associated molecular patterns after an injury such as stroke can lead to the production of large quantities of pro-inflammatory cytokines by microglia, with negative effects on neuronal cells and neurogenesis (Xiong et al., 2016).
Recently, Liu et al. (2019) demonstrated that argon treatment could mitigate brain damage, reduce excessive microglial activation, and subsequently attenuate brain inflammation.We therefore hypothesized that argon might exert its neuroprotective effectsin vitroandin vivoby modulating TLR downstream transcription factors, such as NF-κB, STAT3,activator protein 1 (AP-1), and nuclear factor erythroid 2-related factor 2 (Nrf-2), thereby altering microglial activation and suppressing inflammatory cytokine release.
Materials and Methods
Reagents
Rotenone was diluted in dimethyl sulfoxide to a concentration that would not exceed 0.5% in the cell culture medium.Argon was obtained as a mixture of argon (75%), oxygen(21%), and CO2(the balance was nitrogen) from Air Liquide(Kornwestheim, Germany). The TLR2 and TLR4 inhibitor OxPAPC (Cat# tlrl-oxpap1), rotenone, and dimethyl sulfoxide were supplied by Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and treatments
The cells of a neuroblastoma cell line (SH-SY5Y; Cat# CRL-2266, ATCC, Middlesex, UK) were cultured in a humidified 5% CO2environment at a temperature of 37°C in Dulbecco’s modified Eagle’s medium (DMEM/F12; GIBCO Thermofisher,
Darmstadt, Germany) containing 1% penicillin/retomycin and 10% fetal calf serum. The cells were grown to 80% confluence and then 1.5 × 105cells were seeded in each well of a 6-well culture plate. After 48 hours of culture, the medium was changed to one containing 1% fetal calf serum to ensure that rotenone would not be inactivated by protein binding. The cells were treated with rotenone (20 µM) for 4 hours and then immediately exposed for 2 hours either to room air or to the 75% argon gas mixture in a hermetically sealed box. In TLR2 and TLR4 inhibition experiments, OxPAPC (30 µg/mL) was administered 60 minutes before the rotenone treatment. In all experiments, the cells were harvested immediately after the argon exposure for further analysis.
Transcription factor-binding enzyme-linked immunosorbent assay
The DNA binding activity of transcription factors STAT3, NFκB, Nrf-2, and AP-1 family was determined by DNA binding ELISA according to the manufacturer’s instructions (STAT3:Cat# 45196, NF-κB: Cat# 40096, Nrf-2: Cat# 50296; AP-1 family: Cat# 44296 TransAM®Kit, ActiveMotif, Rixensart,Belgium). Neuroblastoma cells were seeded in 100 mm dishes and incubated until sufficient colonization was achieved.The cells were washed with ice-cold PBS, scraped from the dish surface, centrifuged, and incubated with a hypotonic buffer. Nonidet P-40 was added and the cell lysis was checked under a microscope. After centrifugation and removal of the supernatant, a lysis buffer was added. The protein content of the lysates was determined by the Bradford method (Bradford,1976). Samples containing 10 µg protein were then incubated with the primary antibodies for 1 hour, and the DNA binding activity of the transcription factors was analyzed by measuring the optical density at 450 nm on a microplate reader (Tecan®,Männedorf, Switzerland).
Flow cytometry of the Fas ligand
The cells were washed several times with cold phosphatebuffered saline and then trypsinized and resuspended in 100µL binding buffer. Staining was initiated with a fluorescein isothiocyanate (FITC) conjugated anti-Fas ligand (FasL)antibody (Abcam, Cambridge, UK, Cat# ab10511; 1:40) and the cells were incubated for 15 minutes at room temperature.A 400 µL volume of binding buffer was then added and flow cytometry was conducted using an acoustic focusing cytometer (Attune®, Life Technologies, Darmstadt, Germany).Background fluorescence was determined by compensation and gate setting; unstained cells were used as negative controls.
Animals
Adult male and female Sprague-Dawley rats (male-to-female ratio 1:1, 280-350 g body weight; Charles River, Sulzfeld,Germany) were housed in a 12-hour light/12-hour dark cycle and fedad libitumwith a standard diet. All procedures performed on the animals were in accordance with the statement of The Association for Research in Vision and Ophthalmology for the use of animals in research and were approved by the Commission for Animal Care of the University of Freiburg (approval No. 35-9185.81/G14-122) on October 19, 2012. All interventions were performed as described previously (Ulbrich et al., 2014, 2015b). Animals were randomized to three groups: I ischemia/reperfusion (n= 6), II ischemia/reperfusion+argon (n= 6), III ischemia/reperfusion+argon+OxPAPC (n= 6).
The number of animals used for molecular analysis wasn= 6 per group. The number of animals used for immunoperoxidase labeling wasn= 6 per group. Enucleation was performed after 24 hours.
Retinal ischemia/reperfusion injury and argon treatment
The rats were subjected to intraperitoneal anesthesia[ketamine 10% (80-100 mg/kg) and xylazin 2% (5-10 mg/kg)]and then the anterior chamber of the left eye was cannulated with a 30-gauge needle. A reservoir containing 0.9% NaCl was used to increase the intraocular pressure to 120 mmHg for a period of 60 minutes (Biermann et al., 2010; Ulbrich et al.,2014, 2015c). Retinal ischemia was verified with an optical microscope (Stemi DV4 SPOT, Carls Zeiss, Jena, Germany) as interruption of the retinal blood flow. The neuroprotective effect of argon was determined by randomizing the animals and treating them either with room air or with the argon mixture (argon 75%, oxygen 21%, balance nitrogen) in an airtight chamber for 60 minutes. Post-conditioning was initiated immediately after retinal ischemia (ischemia/reperfusion injury). In TLR2 and TLR4 inhibition experiments,OxPAPC (1.5 mg/kg) was administered intravenously via the tail vein 60 minutes before retinal ischemia.
Real-time polymerase chain reaction
Total RNA was extracted from one fourth of the retinal tissue harvested 24 hours after ischemia using a column purificationbased kit (RNeasy Micro Kit, Qiagen, Hilden, Germany)according to the manufacturer´s instructions. For cell cultures,approximately 2 × 106cells were washed, trypsinized, and processed, and 50 ng of total RNA was used for reverse transcription using random primers (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Darmstadt,Germany). The real-time polymerase chain reaction was performed with the TaqMan®probe-based detection kit(TaqMan®PCR universal mastermix, Applied Biosystems) with the following prímers: interleukin (IL)-1α Rn00566700_m1, IL-1β Rn00580432_m1, IL-6 Rn01410330_m1, tumor necrosis factor α (TNF-α) Rn01525859_g1, eNos Rn02132634_s1, and inducible nitric oxide synthase (iNOS) Rn00561646_m1 (all from Applied Biosystems). PCR was conducted with a RT-PCR system (StepOneTM, Applied Biosystems) and the following parameters: 95°C for 10 minutes, 40 cycles at 95°C for 10 seconds, and 60°C for 1 minute. Reaction specificity was tested with appropriate negative controls. Subsequently, the cycle threshold values (CT) for each gene were normalized to the corresponding CT values for GAPDH (ΔCT). The relative gene expression of the ischemia/reperfusion injured retinal tissue exposed to argon or room air was then calculated in relation to the corresponding gene expression of the noninjured retinal tissue of each individual animal (ΔΔCT).
Immunoperoxidase labeling
After fixation, immunoperoxidase labeling was performed as previously described (Noailles et al, 2014). Shortly after enucleation, the retinas were prepared and placed flat on a nitrocellulose filter with the photoreceptor layer facing up. The samples were placed in 1% H2O2(Sigma) and then incubated in 2.28% sodium metaperiodate (Sigma) and 0.02% sodium borohydride (Sigma). After blocking in 10% normal goat serum for 1 hour, the retinas were incubated in PBS plus 0.5% Triton X-100 for 3 days at 4°C under stirring with the primary antibody mouse-anti-rat integrin-alpha M (CD11b) clone OX-42 (1:500;Cat# 1512; Chemicon, Temecula, CA, USA). After a washing step, the samples were incubated for 24 hours at 4°C in biotinylated goat-anti-rabbit secondary IgG antibodies (dilution 1:100). After washing, the retinas were immersed in a solution of avidin-biotin-peroxidase complex (ABC) (Elite ABC-Kit, Vector Laboratories, Cambridgeshire, UK). The retinas were then pre-incubated with 3,3′-diaminobenzidine tetrahydrochloride(DAB, Sigma; 0.5 mg/mL in PBS) for 10 minutes in the dark under stirring, followed by incubation with fresh DAB solution containing 0.033% H2O2and 0.025% ammonium nickel(II)sulfate hexahydrate (Sigma), according to the manufacturer’s instructions. The DAB reaction was stopped by washing with distilled water. All the retinas were placed flat in Citifluor(Citifluor, London, UK) and covered for optical microscopy(Stemi DV4 SPOT, Carls Zeiss, Jena, Germany).
Immunohistochemical staining
Ionized calcium binding adaptor molecule 1 (Iba-1) is a microglial/macrophage-specific calcium-binding protein.Iba-1 expressing cells in the retina were identified by immunohistochemistry. The rat eyes were enucleated (n= 6 per group) 24 hours after ischemia/reperfusion injury. The retinal tissue was removed and placed as a whole mount preparation in ice cold Hank’s balanced saline solution.The retinas were carefully transferred to a nitrocellulose membrane with the ganglion cell layer on top. The vitreous body was then removed and the specimens were fixed in 4% paraformaldehyde for 1 hour. Immunolabeling was performed according to standardized protocols (Trivino et al., 2002; Rojas et al., 2014) with mouse monoclonal antibody against Iba-1 (Cat# ab15690; dilution 1:150;Abcam). Conjugation of the primary antibody with the corresponding secondary antibodies was shown as follows.A green fluorescence was generated with Alexa Fluor 488(goat anti-mouse, dilution 1:1000; Cat# 115-545-003,Jackson ImmunoResearch Europe, Cambridgeshire, UK). The nuclei of cells in the retina were stained with 4’,6-diamidino-2-phenylindole-dihydrochloride hydrate (DAPI, Sigma,Taufkirchen, Germany). The preparations were covered with embedding medium (Mowiol; Calbiochem, San Diego, CA,USA) and stored at 4°C.
Analysis of microglia
Three-dimensional confocal laser scanning microscopy overview and close-up images were obtained with a LSM 510 device (Carl Zeiss, Jena, Germany) at 20× and 40×magnification, respectively. Z-stack images were deconvoluted using HuygensEssential 1605p5 software (SVI, Hilversum,The Netherlands). For representative purposes, all images from each Z-stack were either focal plane merged (overview images) or 3D reconstituted (close-up images) using Imaris 8.3.0 image analysis software (Bitplane, Zurich, Switzerland).Quantification and histogram analysis of immunostained images were performed using the ImageJ open source software (ImageJ, Version 2.00-rc-44/1.50e, https://imagej.nih.gov/ij/plugins/fraclac/FLHelp/Introduction.htm). Microglial morphology in relation to the cell shape was determined by measuring the density (Morrison et al., 2017) using the FracLac plug-in from ImageJ. This generates a “convex hull,”where each microglia is surrounded by a polygon and a circle that borders the convex hull. The density is then the ratio of the number of pixels covered by the cell outline to the area (in pixels) of the convex hull.
Statistical analysis
A computerized statistics program (SigmaPlot Version 11.0,Systat Software Inc., San Jose, CA, USA) was used to evaluate the data. After checking the data for a normal distribution,the results were presented as mean values (SD). Betweengroup comparisons were performed with the one-way analysis of variance followed by thepost-hocHolm Sidak test (or the Kruskal-Wallis test for data without a normal distribution). A value ofP< 0.05 was considered statistically significant.
Results
Argon reduces binding activity of STAT3 and NF-κB in SH-SY5Y cells exposed to rotenone
Exposure of human neuroblastoma cells (SH-SY5Y) to rotenone,a substance that blocks the mitochondrial respiratory chain and induces apoptosis, significantly increased STAT3 and NF-κB binding activity (P< 0.001;Figure 1AandB). Exposure to argon for another 2 hours caused a partial reduction in the STAT3 and NF-κB activity (P< 0.05 andP< 0.001;Figure 1AandB). Argon alone, in the absence of rotenone pretreatment, had no effect on STAT3 and NF-κB activity, while OxPAPC treatment reversed the argon effect in rotenone-treated cells (P< 0.05 andP<0.001;Figure 1AandB).
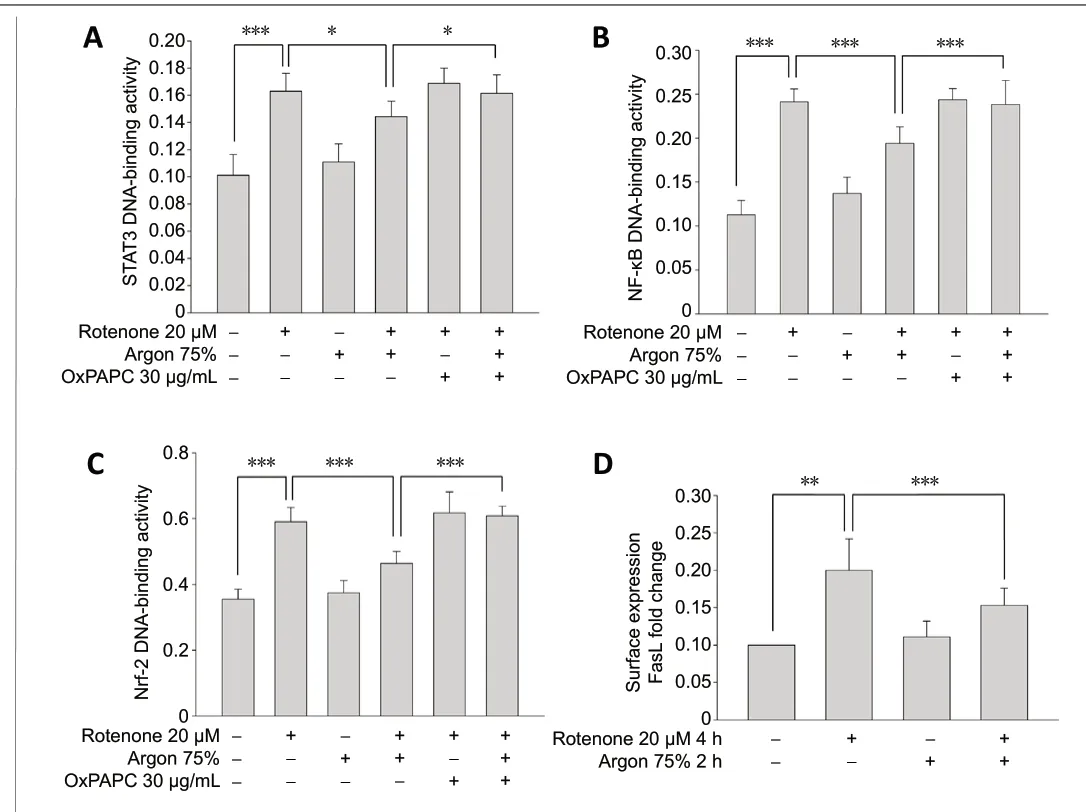
Figure 1 |Argon reduces STAT3, NF-κB, and Nrf-2 DNA-binding activity and FasL surface expression in SH-SY5Y cells exposed to rotenone.(A, B) Argon (75%, 2 hours) reduced STAT3 and NF-κB DNA-binding activity of human neuroblastoma cells (SH-SY5Y) after 4-hour rotenone (20 µM)pretreatment. STAT3 and NF-κB showed an increased binding activity after ischemia/reperfusion injury, which could be significantly reduced by argon or almost completely eliminated, as in the case of NF-κB. Argon alone, in the absence of rotenone pretreatment, had no effect. Previous application of the TLR2 and TLR4 inhibitor oxidized phospholipid 1-palmitoyl-2-arachidonoylsn-glycero-3-phosphorylcholine (OxPAPC, 30 µg/mL) had no effect on cells treated with rotenone. However, OxPAPC neutralized the effect of argon after ischemia/reperfusion injury. Argon decreased Nrf-2 DNA binding activity (C).Argon decreased Nrf-2 DNA binding activity. DNA binding enzyme-linked immunosorbent assay of Nrf-2 activity after rotenone treatment and argon(75%, v/v, 2 hours) fumigation. OxPAPC abolished Argon’s effect. Argon reduced FasL surface expression (D). Argon reduced FasL surface expression(fluorescence-activated cell sorting analysis). n = 6 in A-C; n = 8 in D. Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (oneway analysis of variance with post-hoc Holm Sidak test). FasL: Fas ligand; NFκB: nuclear factor kappa B; Nrf-2: nuclear factor erythroid 2-related factor 2;STAT3: signal transducer and activator of transcription 3.
Argon reduces binding activity of Nrf-2 in SH-SY5Y cells exposed to rotenone
Treatment of SH-SY5Y cells with rotenone significantly increased Nrf-2 binding activity (P< 0.001;Figure 1C), while subsequent treatment with argon mitigated rotenone effect and decreased Nrf-2 transcriptional activity (P< 0.001;Figure 1C). Argon alone, in the absence of rotenone pretreatment,showed no impact on Nrf-2 DNA binding. OxPAPC treatment almost completely eliminated the effect of argon in rotenonetreated cells (P< 0.001;Figure 1C).
Argon decreases FasL surface expression in SH-SY5Y cells exposed to rotenone
The possible effect of argon on FasL expression was evaluated by fluorescence-activated cell sorting (FACS) analyses. In SH-SY5Y cells, exposure to rotenone increased the surface expression of FasL (P< 0.01;Figure 1D). Immediate argon treatment reduced the effect of rotenone and diminished FasL surface density, while argon alone, in the absence of rotenone pretreatment, showed no effect (P< 0.001;Figure 1D).
Argon differentially inhibits AP-1 proteins in SH-SY5Y cells exposed to rotenone
Rotenone pretreatment of SH-SY5Y cells did not change the DNA binding activity of JunD and c-Fos (Figure 2AandB), but significantly increased binding of Fra-1 and c-Jun (P< 0.001;Figure 2CandD), indicating differential activation of AP-1 transcription proteins. However, the increase in FosB and JunB activity did not reach statistical significance (Figure 2EandF). Argon treatment alone, in the absence of rotenone pretreatment, had no impact on AP-1 protein binding (Figure 2A-F). However, OxPAPC treatment did not reverse the argon effect on DNA-binding activity for Fra-1 and c-Jun in rotenonetreated cells (Figure 2CandD).

Figure 2 |Argon differentially regulates AP-1 proteins in SH-SY5Y cells exposed to rotenone.(A-F) DNA binding activity of JunD, c-Fos, Fra-1, c-Jun, FosB, and JunB(enzyme-linked immunosorbent assay). n = 6. Data are expressed as the mean± SD. ***P < 0.001 (one-way analysis of variance with post-hoc Holm Sidak test). AP-1: Activator protein 1; n.s.: not significant; OxPAPC: the TLR2 and TLR4 inhibitor oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine.
Argon inhibits thein vivorelease of inflammatory cytokines
Retinal ischemia/reperfusion injury in the rats increased the retinal mRNA expression of IL-1α, IL-1β, IL-6, TNF-α, and iNOS compared to untreated control eyes (Figure 3A-F). A 2-hour exposure to argon immediately after reperfusion significantly decreased the ischemia/reperfusion injury-mediated proinflammatory cytokine expression significantly (P< 0.05 orP<0.01;Figure 3A-E). Pretreatment with OxPAPC partly abolished argon’s effect on pro-inflammatory cytokine expression (P<0.05 orP< 0.01;Figure 3A-E). Retinal ischemia/reperfusion injury had no effect on eNOS mRNA expression.
Argon attenuatesin vivomicroglial activation
Immediately after ischemia/reperfusion injury,immunohistological staining with Iba-1 and the subsequent 3D reconstruction of the images revealed an increased number of microglial cell bodies. The cell bodies were also thickened and showed an asymmetric distribution of the extensions, indicative of microglial activation (Figure 4AandB). Immunoperoxidase labeling also showed the typical morphological changes. The density of the microglia increased after ischemia/reperfusion injury, but this change was significantly reduced by argon treatment (Figure 5AandB). OxPAP C abolished the effect of argon (Figure 4CandD;Figure 5AandB;P< 0.05 andP<0.001).
Discussion
Thesein vitroandin vivoexperiments validated the antiinflammatory properties of argon. Argon treatment of human neuroblastoma cells injured by rotenone (1) reduced the binding activity of the transcription factors STAT3, NF-κB, and Nrf-2, (2) caused differential effects on the subunits of the transcription factor AP-1, and (3) and diminished the surface expression of FasL, a cytokine that induces apoptosis.In vivotreatment of rat retinas with argon reduced the activation of retinal microglial cells after ischemia/reperfusion injury,with subsequent suppression of retinal mRNA expression of inflammatory cytokines and iNOS. These effects could be reversed by inhibiting TLR2 and TLR4 signaling with OxPAPC.
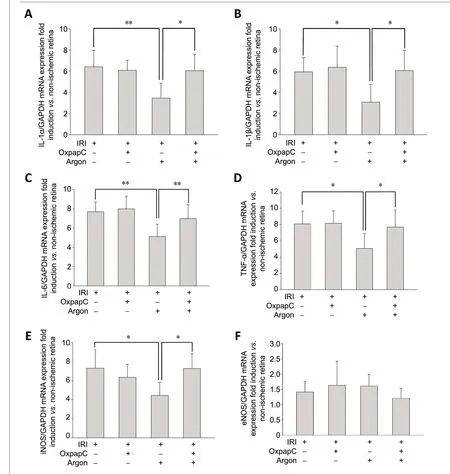
Figure 3 |Argon attenuates retinal ischemia/reperfusion injury (IRI)-mediated mRNA expression of various pro-inflammatory cytokines in rats.(A-F) Analysis of retinal IL-1α, IL-1β, IL-6, TNF-α, iNOS, and eNOS mRNA expression using quantitative polymerase chain reaction. Fold induction in retinal tissue compared to GAPDH in relation to the corresponding nonischemic retinas after treatment with and without argon (75%) and the TLR2 and TLR4 inhibitor oxidized phospholipid 1-palmitoyl-2-arachidonoyl-snglycero-3-phosphorylcholine (OxPAPC, 1.5 mg/kg). n = 6. Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01 (one-way analysis of variance with post-hoc Holm Sidak test). eNOS: Endothelial nitric oxide synthase; IL:interleukin; iNOS: inducible nitric oxide synthase; n.s.: not significant; TNF-α:tumor necrosis factor alpha.
Different neuronal injury models have shown that the destruction of neuronal tissue leads to a release of calcium and reactive oxygen radicals, which in turn stimulate the expression of different transcription factors, such as STAT3,NF-κB, and AP-1, to regulate the expression of proteins responsible for either cell survival or cell death (Chen et al., 2011). Inhibition of the expression of the transcription factor Nrf-2 has also been associated with neuroprotective mechanisms, so Nrf-2 has become a focus in studies regarding stroke and other neurological diseases (Jiang et al., 2017).
Transcription factors are involved in the development of cerebral inflammation and contribute to secondary neurological injury. Microglia, as the resident macrophages of the brain, are responsible for monitoring the microenvironment, so they would be expected to undergo alterations in transcription factors. Microglia undergo activation following neuronal damage, with a subsequent release of inflammatory cytokines. In the activated state, the number of microglia cells increase, with concomitant changes in their morphology. The cell body thickens and assumes an amoeboid shape, with an asymmetrical distribution of the processes (Chen and Trapp, 2016).
In previous experiments, we have shown that microglial activation is alleviated by argon treatment through effects on TLR2 and TLR4, which play important roles in neuroprotection(Ulbrich et al., 2015b, 2016). The presentin vitroexperiments in rotenone-treated human neuroblastoma cells confirmed that argon reduced the binding activity of TLR downstream transcription factors, such as STAT3, NF-κB, and Nrf-2, thereby preventing the neuronal cell death induced by rotenone inhibition of the mitochondrial respiratory chain. We further confirmed this effect on Toll-like receptors using OxPAPC to inhibit TLR2 and TLR4. OxPAPC alone was not neurotoxic,but OxPAPC treatment reversed the beneficial effects of argon on rotenone-treated cells. By contrast, the activity of transcription factor AP-1 was only partly inhibited and was not antagonized by OxPAPC, suggesting the involvement of still yet another mechanism.
Some studies have shown an association between neuronal markers and anti-apoptotic proteins, like microtubuleassociated protein 2 and BCL-2, which would suggest a correlation with neuronal survival (Zhang et al., 2006; Xiong et al., 2011). By contrast, other studies have linked STAT3 activation to neuronal cell death (Dziennis and Alkayed,2008). A recent study by Ito et al. (2019) on the influence of regulatory T cells on ischemic brain damage revealed that stroke induced STAT3 activation, but this activation was reduced by intraventricular injection of the neuroprotective amphiregulin released by regulatory T-cells. A correlation between neuroprotection and downregulation of NFκB in association with Toll-like receptors has also been demonstrated in the several studies. For example, Hwang et al. (2017) showed that NF-κB provided neuroprotective effects via TLR4 in rats after cerebral ischemia and post-conditioning with sevoflurane. Similarly, Nalamolu et al. (2018) confirmed the contribution of TLR2 and TLR4 in focal cerebral ischemia in rats, by knockdown of TLR2 and TLR4. This knockdown prevented the upregulation of downstream pathways,including NF-κB, and suppressed the subsequent release of inflammatory cytokines, thereby reducing infarct size and swelling.
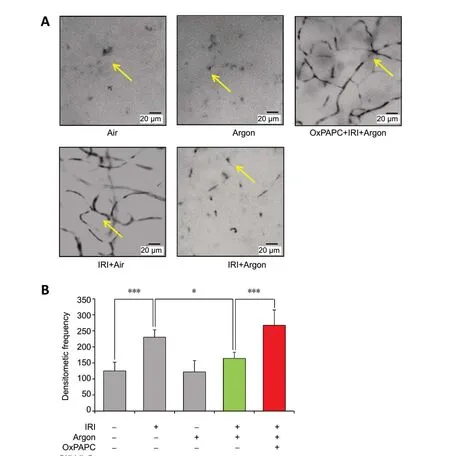
Figure 5 |Microglial activation in whole-mount specimen after immunoperoxidase labeling with CD11b (OX-42).(A) Visualization of the morphology of retinal microglia by optical microscopy:Control eye, after ischemia/reperfusion injury (IRI), after IRI and argon postconditioning, and after IRI treatment after application of the TLR2 and TLR4 inhibitor oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (OxPAPC, 1.5 mg/kg). Typical changes such as increase of microglial cell bodies, thickening and asymmetric distribution of extensions showing activation after ischemia/reperfusion, which is attenuated by argon(arrows). (B) Statistical analysis of microglial density. Argon significantly reduced the density increase due to IRI. Argon alone had no effect in the absence of IRI treatment. The previous application of OxPAPC prior to IRI neutralized the effect of argon. n = 6. Data are expressed as the mean ± SD.*P < 0.05, ***P < 0.001 (one-way analysis of variance with post-hoc Holm Sidak test).
Nrf-2 is an important element in the protection against neuronal inflammation, as it controls the release of antioxidant substances and influences the activation of microglia (Lee et al., 2005). Our study showed a significant reduction in Nrf-2 in response to argon exposure. Argon is viewed as a potent mediator of organ protection, as it upregulates the expression of a variety of antioxidant enzymes during oxidative stress. Our finding is contradictory to that of Zhao et al. (2016), who found an activation of Nrf-2 during argon-mediated neuroprotection after hypoxic-ischemic brain damage. The reason for this difference may be that Zhao et al. supplied argon over a period of 24 hours after oxygenglucose withdrawal, and they processed and analyzed the cells after that time. By contrast, our protocol involved rotenone treatment for 4 hours, argon exposure for another 2 hours,and immediate processing of the cells. We speculate that the decisive factor may be the time interval and that Nrf-2 activation might only be detected at a later point in time after neuronal injury.
Ourin vitrofindings confirmed a contribution of some of the subunits of the AP-1 transcription factor in neuroprotection.JunD, C-Fos, FosB, and JunB showed no changes in response to rotenone, whereas Fra-1 and c-Jun were activated, and argon exposure significantly attenuated their DNA binding activity.Interestingly, OxPAPC could not reverse this effect of argon,suggesting the involvement of yet another mechanism that is independent of the toll-like receptors. The current literature indicates an involvement of proteins of the AP-1 group in the processes of neurodegeneration and neuroregeneration. In particular, c-Jun, a product of the mitogen-activated protein kinase JNK, is stimulated in apoptotic neuronal processes,whereas its inhibition is associated with neuroprotection,according to numerous studies (Herdegen and Waetzig,2001). Experiments on a rat stroke model have confirmed the contribution of the JNK/c-Jun pathway to cerebral ischemia/reperfusion injury by demonstrating an increase in the apoptosis-inducing cytokine FasL, which is a central component of the JNK-pathway (Qi et al., 2010). Transcription factors play a crucial role in the activation of cerebral microglia, as indicated by strong induction by STAT3 (Planas et al., 1996) and pro-inflammatory pathways, such as NF-κB (Lee,2013) and AP-1 (Hui et al., 2018), as well as involvement of the antioxidant regulator Nrf-2 (Vilhardt et al., 2017).
Ourin vivoexamination of retinal microglia confirmed their activation by retinal ischemia/reperfusion damage due to elevated intraocular pressure. The increased number of retinal microglial cells detectable by immunoperoxidase staining was one sign of activation. The immunohistological staining with Iba-1 also revealed morphological hallmarks of activation in the form of thickening of the cell bodies and an asymmetric distribution of the extensions. This microglial activation would be expected to increase the release of proinflammatory cytokines, such as IL-1β, IL-6, TNF-α, and iNOS.
The activation of retinal microglia by ischemia/reperfusion was significantly reduced by argon inhalation. This neuroprotective effect of argon has now been demonstrated in a number ofin vitroandin vivomodels, but the mechanism of action has not yet been fully clarified. Our previous studies demonstrated an effect of argon on TLR2 and TLR4, which are receptors of the innate immune system, and subsequently on the mitogenactivated protein kinase ERK1/2, as well as a downregulation of the pro-inflammatory cytokine interleukin-8. In 2012,Fahlenkamp et al. reported thatin vitroargon exposure increased ERK1/2 phosphorylation in the BV-2 microglial cell line. However, argon had no significant effect on ERK1/2 when the cells were pretreated with lipopolysaccharide;instead, argon treatment increased the release of the proinflammatory cytokine IL-1β. Zhuang et al. (2012) also demonstrated beneficial effects of argon afterin vitrodamage to cortical neuronal cells by oxygen glucose deprivation,as argon exposure attenuated cell death and reduced the expression of the pro-inflammatory cytokines TNF-α and IL-6.Fahlenkamp et al. (2014) showed that argon had noin vivoeffect on the number of microglial cells in the rat during a temporary occlusion of the middle cerebral artery, although further ramifications on microglia were not investigated.However, expression analysis following argon exposure showed only a reduction in CD3 by argon and even an increase in the inflammatory cytokines IL-1β and IL-6. A recent study by Liu et al. (2019) suggested that argon suppression of microglial activation in a rat stroke model may be due to a promotion of M2 microglia/macrophage polarization, resulting in an anti-inflammatory macrophage phenotype.
In the present study, ischemia/reperfusion injury caused morphological alterations of retinal microglia in the form of thickened extensions, and it increased the numbers of retinal microglia cells. Argon exposure, in turn, reduced both the morphological changes and the density of retinal microglial cells, suggesting a reversal of activation. Moreover,argon reduced the release of iNOS, a marker of the proinflammatory microglial phenotype (Collmann et al., 2019).
Conclusion
The results of thisin vitroandin vivostudy provide further details of the mechanism underlying argon neuroprotection.Argon appears to exert its neuroprotective effect by attenuating a number of transcription factors associated with toll-like receptors, thereby reducing microglial activation.Argon is inexpensive and apparently has no adverse side effects, so its use in humans is feasible. Clinical studies should therefore be pursued. In addition, growing evidence supports the use of argon to benefit other organ systems, such as the heart and kidneys.
Acknowledgments:The authors thank Heide Marniga from Department of Anesthesiology and Critical Care, Medical Center - University of Freiburg(medical-technical assistant) for the support. The authors also thank the staff of the Live Imaging Center Freiburg for the assistance collecting and evaluating images.
Author contributions:UG designed the study, is responsible for data analysis and evaluation, and wrote the manuscript. SSc conducted the experiments and helped with data analysis and evaluation. SSp conducted the experiments,and helped with data analysis and evaluation. NS and JW helped with data analysis and evaluation, and edited the manuscript. HB helped with data analysis and evaluation, and was an advisor in the management of the study.FU designed the study, conducted the experiments, helped with data analysis and evaluation, and wrote the manuscript. All authors approved the final version of this manuscript.
Conflicts of interest:There are no competing interests .
Financial support:This work was financially supported by the Department of Anesthesiology and Critical Care, Medical Center - University of Freiburg,Germany. The article processing charge was funded by the Baden-Württemberg Ministry of Science, Research and Art and the University of Freiburg in the funding programme Open Access Publishing.
Institutional review board statement:All procedures involving animals concurred with the statement of The Association for Research in Vision and Ophthalmology for the use of animals in research in accordance with the Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines and were approved a priori by the Committee of Animal Care of the University of Freiburg (approval No. 35-9185.81/G14-122) on October 19, 2012.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study areavailable from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Information for Authors-Neural Regeneration Research
- Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke
- Laminin-coated multifilament entubulation, combined with Schwann cells and glial cell line-derived neurotrophic factor, promotes unidirectional axonal regeneration in a rat model of thoracic spinal cord hemisection
- Nogo-A aggravates oxidative damage in oligodendrocytes
- Insights into stem cell therapy for diabetic retinopathy:a bibliometric and visual analysis
- Deletion of Krüppel-like factor-4 promotes axonal regeneration in mammals