
满江红内生细菌的培养及其抗生素抗性基因的检测
2021-08-18 06:50:54郑益平郑斯平郑伟文
福建农业学报 2021年6期
陈 坚,郑益平,陈 彬,郑斯平,郑伟文
(福建省农业科学院生物技术研究所,福建 福州 350003)
0 引言
【研究意义】发明抗生素类药物治疗疾病是人类在20 世纪取得的最重要成就之一。但是由于微生物的变异迅速,引入抗生素后不久它们就会出现抗性基因ARGs(Antibiotic resistance genes),使治疗药物失效。并且在任何一种新的抗菌药物引入后,都可能产生新的抗性基因,从而造成对抗生素的耐药性[1]。2017 年,Razavi 等[2]报道,ARGs 可以通过可移动遗传原件(Genetic mobile elements)的机制在细菌中传播,造成ARGs 在环境中扩散,从而对公共健康构成威胁。由于在水产养殖的实践中,抗生素不仅具有防治水产动物细菌性疾病的效能,还能起到促进养殖动物生长的作用,因此在水产养殖业中的使用极为广泛[3-4]。滥用抗生素导致抗生素抗性基因在水源环境中迅速传播,不仅对水产养殖业的发展造成直接危害,也对人类健康产生重要影响[5-7]。因此,监测和鉴定抗生素抗性基因在水源环境中的动态成为重要的课题。【前人研究进展】在水源环境中广泛存在的水生蕨类植物满江红(Azolla)俗称红萍、绿萍,因富含氮素和蛋白质等营养成分,在亚洲农村被用作绿肥和饲料已有数百年历史[8-9]。这种细小的植物繁殖快,生物量高,归因于其叶腔中栖息着一种固氮蓝藻。该蓝藻形态酷似念珠藻(Nostoc),一般称为满江红念珠藻(Nostocazollae),是由多个细胞串联成的丝状体,含有营光合作用的营养细胞和专司固氮的异形胞。异形胞含有编码固氮酶的nif基因,可将大气氮转化为植物可利用的结合态氮,以NH4的形态满足宿主生长对氮素的需求。宿主则以蔗糖为主的碳源作为回报[10-11]。但近30 年来,人们先后从满江红的叶腔发现并分离培养出其第三个共生伙伴——细菌。它们与固氮蓝藻一起构成了满江红叶腔内的微生态系统。研究者们报道以自然界的满江红作为研究满江红内生细菌的材料,采用传统的平板培养法,从4 个生物种的满江红中分离鉴定出9 个属15 个种的内生细菌[12-13]。进入21 世纪后,随着分子生物学技术的进步,美国Raensallae 大学的Lechno 等[14]用rDNA-PCR 等方法从卡洲满江红的叶腔中鉴定出根癌农杆菌、根瘤菌等4 种细菌。郑斯平等[15]用PCRDGGE 和电镜技术对满江红的叶腔和孢子果内的细菌进行了检测和观察,揭示了小叶满江红叶腔以变形菌门为主的多种可培养和不可培养的细菌,把对满江红内生菌的表型多样性的研究推进了一步。随后,郑斯平等[16]将小叶满江红的内生菌经16S rDNA-PCR扩增后用T-RFLP 技术和PAT 软件分析,通过比对确定这些内生菌属于10 科42 属。【本研究切入点】鉴于满江红是一种在淡水环境中生长的古老植物,内生有丰富的微生物群落。鉴定与检测满江红内生菌的抗生素相关基因,可以从一个侧面了解ARGs在水环境中传播的状况。【拟解决的关键问题】常用于检测环境中抗生素抗性基因ARGs 的技术是PCR和微阵列技术,但近年来宏基因组学(Metagenomics)是新发展的一种基因组学技术,它通过从环境样品中提取全部微生物的DNA,构建基因组文库,利用基因组学的方法研究环境中所包含的全部微生物的基因组信息,以探究微生物的群落结构和基因功能。随着新一代测序技术的完善,以及测序成本的逐年快速下降,基于宏基因组学的抗生素抗性基因ARGs 的鉴定逐渐成为主流技术[17-18],本研究拟用3 种不同的培养基对小叶满江红内生菌进行培养,利用宏基因组学技术对培养的内生细菌进行群落结构及抗生素抗性基因的分析和检测,为进一步研究满江红内生细菌的存在状态,以及利用内生细菌中抗生素抗性基因的检测,了解植物与生长环境的互作关系奠定基础。
1 材料与方法
1.1 满江红植物材料来源与制备
供试材料为小叶满江红(Azolla microphyllaKaulfus),早先从国际水稻研究所(International Rice Research Institute)引进,原编号为IRRI 4018。试验前在网室中于自然气候条件下长期培养保存,培养所用基质为水土培养基,由取自田间的自然土高压灭菌后加自来水配制而成。试验时为了排除外源细菌等微生物的干扰,参照白克智等[19]的茎尖培养法,略为修改进行材料预处理,具体如下:取100 g(鲜重)在自然条件下生长的萍体,用自来水清洗,去掉黏附的土壤等杂物,剪去根系。后选取健康的萍体再用无菌水清洗。捞取洗净的健康萍体,置于灭菌的烧杯中,加入适量的0.1%升汞溶液,将萍体浸没,用灭菌的镊子连续搅拌3.5 min,使满江红的叶、茎和幼根充分与溶液接触,以确保灭菌效果。随后倾尽升汞溶液,迅速用无菌水清洗经灭菌的萍体,至少清洗4 次,每次3 min,尽量洗去残留在萍体上的升汞。在立体解剖镜下用无菌的细针挑取并弃去包被茎尖的叶片,仅保留茎尖分生组织和1~2 个叶原基,后投入灭菌的IRRI培养液的三角瓶内。IRRI培养液的配方为(mg·L-1):NaH2PO4·H2O 89,K2SO489.1,CaCl2·2H2O 147,MgSO4·7H2O 405.3,MnCl2·4H2O 1.8,Na2Mo2·2H2O 0.38,H3BO31.14,ZnSO4·7H2O 0.04,CuSO4· 5H2O 0.04,CoCl2· 6H2O 0.04,EDTA-Fe(FeSO4· 7H2O 0.249,EDTA 0.261,KOH 0.157,pH 6.5。
以上操作均在组培实验室的无菌环境下(苏净SW-CJ-2F 型)完成。
培养条件为:26 ℃/18 ℃(日/夜),光强15 000 lx。10~15 d 更新培养液。待繁殖足量后分别称取样品用于下一步试验。
1.2 培养组微生物样品的制备
称取培养的满江红鲜重材料2.5 g,加入6 mL 的ddH2O 研磨至匀浆,而后取1.5 mL 的满江红萍体匀浆液,接种到不同的微生物液体培养基中进行震荡培养,培养温度25 ℃,震荡转速为60 r·min-1,培养时间为7~14 d。本研究选择最通用的2 种细菌培养基R2A[20]及TRN[12]对满江红的内生细菌进行培养,并设置以培养满江红无藻萍的Nickell[19]培养基为对照。满江红内生细菌培养组的培养基具体配方见表1。
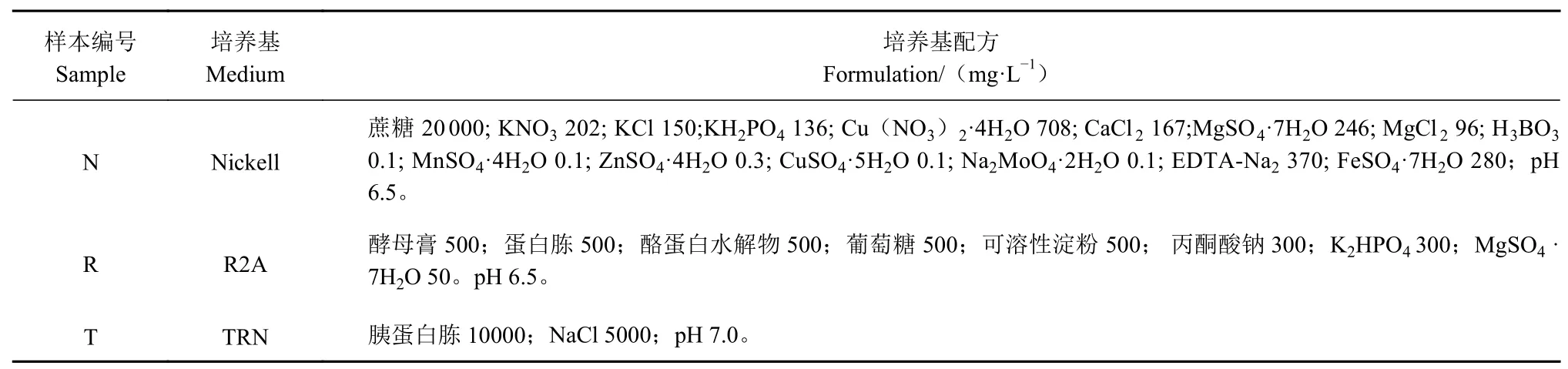
表1 满江红内生菌的培养基Table 1 Culture media for endophytic bacteria of Azolla
1.3 培养组样品DNA 提取与检测
培养1~2 周后的培养组微生物样品,经离心收集菌体,用于全基因组DNA 的提取。本研究采用Ezup柱式细菌基因组 DNA 抽提试剂盒(上海生工),并按说明书的操作步骤进行。提取的DNA 需经过质检,构建IlluminaPE 文库的DNA 含量≥2 ng·μL-1,DNA 质量 ≥0.25 μg。检测前将样品在冰上融化后,充分混匀并离心,取适量样品进行检测。分别用NanoDrop2000 检测 DNA 纯度,QuantusFluorometer(Picogreen)检测DNA 含量以及用1% 琼脂糖凝胶电泳对检测所提取DNA 的完整性(电压:120 V,时间:20 min)。
1.4 宏基因组测序与生物信息学分析
样品的宏基因组测序委托上海美吉生物医药科技有限公司在其Illumina 第二代测序平台MiSeq 上进行,数据分析从下机原始序列开始,首先对原始序列进行拆分、质量剪切以及去除污染等优化处理。然后使用优化序列进行拼接组装和基因预测,对得到的基因比对物种数据库NR,以及抗生素相关基因数据库ARDB 和CARD,进行抗生素抗性基因功能上的注释以及分类。宏基因组的数据分析在美吉生物科技有限公司的生物云平台(i-sanger)上完成。
2 结果与分析
2.1 宏基因组测序结果的统计分析
对满江红内生细菌3 个培养组的宏基因组测序的有关数据见表2。每个序列的读长为150 bp,获得的原始读数为101.34~114.87 M;原始的碱基数为15.30~17.35 G。使用软件fastp 对原始测序数据进行质控,剪切掉数据中的低质量及含N 的reads,从而得到高质量的质控数据(clean data)。获得质控后的读数为99.91~113.81 M,质控的碱基数为15.08~17.18 G,本次宏基因组测序共产生高质量的数据量为48.66 G,质控后碱基覆盖原始碱基数的98.52%以上。
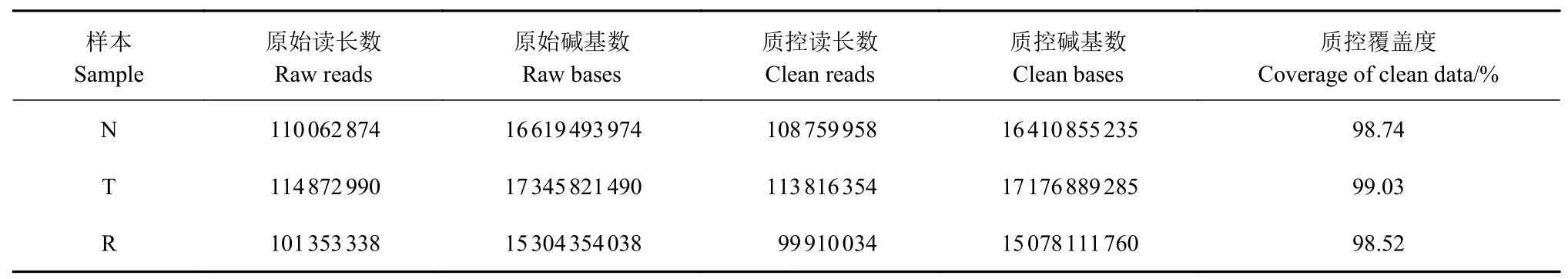
表2 宏基因组测序数据的统计Table 2 Summary of metagenomic sequencing data
在获得质控数据的基础上,对序列进行组装和基因预测,处理结果见表3。将3 个培养组样本所获得的质控读长通过Megahit 进行迭代拼接(overlap),拼接片段(contigs)最短要求为300 bp,各样本得到1 711~13 482 个拼接片段(contigs)。使用MetaGene进行ORF 预测。选择核酸长度大于等于100 bp 的基因,并将其翻译为氨基酸序列。不同样本测得11 813~27 163 个基因,每个样品基因的平均长度623.17~903.83 bp,覆盖数据量为9.16~16.93 M。将预测出来的总共54 180 个基因(ORFs)序列使用CD-HIT 软件进行聚类(参数为:90% 相似度、90% 覆盖度)构建非冗余基因集(Catalog gene),用于生物信息学分析。
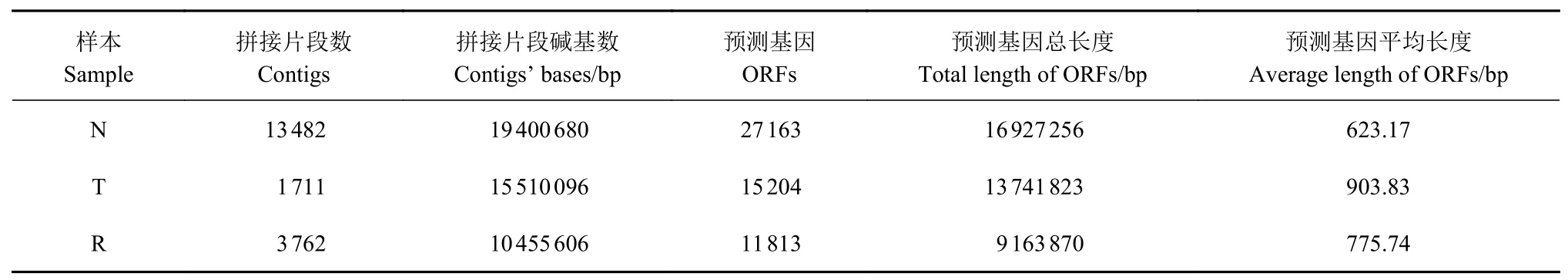
表3 拼接片段与预测的功能基因数据的统计Table 3 Summary of contigs and predicted genes
2.2 培养的内生菌群落结构分析
使用 BLASTP 将非冗余基因集与NR 数据库(RefSeq 非冗余蛋白质的氨基酸序列数据库,https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/)进行比对(BLAST 比对参数设置期望值 e-value 为1e-5),并通过 NR 库对应的分类学信息数据库获得物种注释结果。从所采集的培养样品中,共注释出1 712 种微生物物种,使用物种对应的基因丰度总和计算该物种的丰度,在各个分类学水平上可以统计出物种在各个样品中的丰度。将注释到的样品微生物类别在总门水平和总属水平上进行分类所得到物种及其丰度见表4,在云平台分析仅显示丰度大于0.01阈值的物种名称时,尽管在总门水平共有38 个门的物种被命名,但通过统计物种丰度发现:在所有培养样品中仅变形菌门(Proteobacteria)占比在99.14%以上,其余门占比极低,总丰度仅有0.64%~0.86%。可见满江红内生菌在所选用的3 种培养基中均培养出以变形菌(Proteobacteria)为优势的细菌。
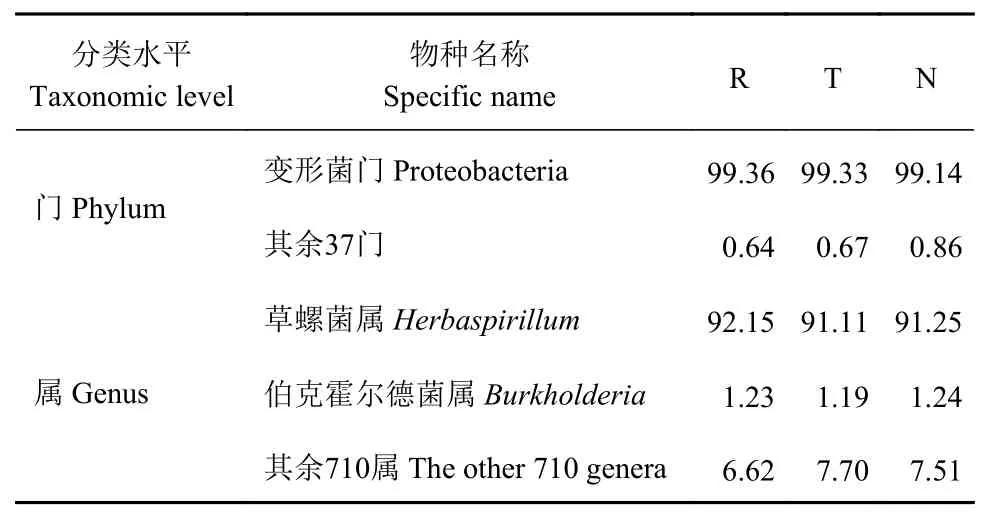
表4 不同样本的微生物在门与属分类水平上的物种丰度Table 4 Microbial community abundance of cultured samples at phylum and genus levels (单位:%)
由表4 可见:3 个样品中虽然可检测出712 个物种属,但其中草螺菌属(Herbaspirillum)的丰度为91.11%~92.15% ;其次为伯克霍尔德菌属(Burkholderia),在各样本中的占比可达1.2%左右。其余属的单属占比均小于1%,合并总量仅占到7%左右。可见在总属水平,3 种所选培养基中满江红内生菌均繁殖出以草螺菌属(Herbaspirillum)为优势的细菌。
2.3 抗生素抗性基因数据库的比对分析
抗生素抗性基因数据库[21](ARDB,http://ardb.cbcb.umd.edu/)收集了不同环境来源(如生活废水、河流等)的细菌抗药性基因及其抗性谱、作用机制等注释信息,为研究药物作用、环境治理提供研究依据。使用 BLASTP 将非冗余基因集与 ARDB 数据库进行比对(比对参数设置期望值 e-value 为 1e-5),3 个样品共注释到10 个与抗生素有关的ARG 基因,归为baca、bcra、bl2a_iii及ceob四大类型(type),涉及杆菌肽(bacitracin)氯霉素(chloramphenicol),和青霉素(penicillin)等3 种抗生素,将样品中的抗性基因的数量与相应的抗生素类型总结于表5。由表5中可见:满江红内生细菌携带的大量抗性基因主要针对2 种抗菌素,一种为杆菌肽,另一种为氯霉素。

表5 样本中抗生素抗性基因的类型与序列读长数Table 5 Types and read amounts of ARGs in cultured samples
综合抗生素抗性基因数据库CARD[22](http://arpcard.Mcmaster.ca,Version 1.1.3)广泛包含了来自各种生物体、基因组、质粒的抗生素抗性相关的参考基因,可用于指导环境、人体、动物菌群耐药组和抗生素抗性机制研究。使用 BLASTP 软件,把非冗余基因集与 CARD 数据库进行比对,比对参数设置为“严格(strict)”比对。将目标物种的基因与其耐药功能注释信息结合,3 个样本中总共注释得到212 个抗生素相关基因ARO(Antibiotic resistance ontology)。这些抗性基因再依据CARD 对基因抗性机制的基本分类,主要对应于3 个类别(Class)水平:第一类为AR(Antibiotic Resistance)类抗生素抗性基因,它们能直接抵抗抗生素的作用,消除抗生素对细胞内环境的影响。3 个样本中这类抗性基因的丰度均占88%以上;第二类为AS(Antibiotic Sensitive)类抗性基因,它们通过发生突变、敲除等方式,而赋予菌株抗生素抗性。这类抗性基因在样本中丰度为7%左右;第三类为AT(Antibiotic Target)类抗性基因,它们是与抗生素作用靶点结合后,能实现抵抗抗生素的作用,这类抗性基因在样本中占4%左右;将CARD 注释的各样本中的抗性基因ARO 的数量和抗性机制类别(Class)的丰度总结于表6 中,由表6可见:3 个样本检测到的抗生素抗性基因的机制类别是基本相同的。不仅如此,其中丰度最高的10 个ARO也基本相同,由表7 可见:3 个样本中丰度最高的ARO 依次为:macB、sav1866、mdtB、PmrB、mexT、PmrA、evgS、adeH、mdtD、rosB。其中PmrA,PmrB为多粘菌素类抗性基因,通过改变细胞壁的负荷抵消抗生素作用,其余8 个都是与通过泵的机制将抗生素外排作用有关的基因。这10 个ARO 均属于AR类抗性基因。由此可见:本研究检测到的满江红内生细菌主要是以直接抵抗的机制以消除抗生素的作用。
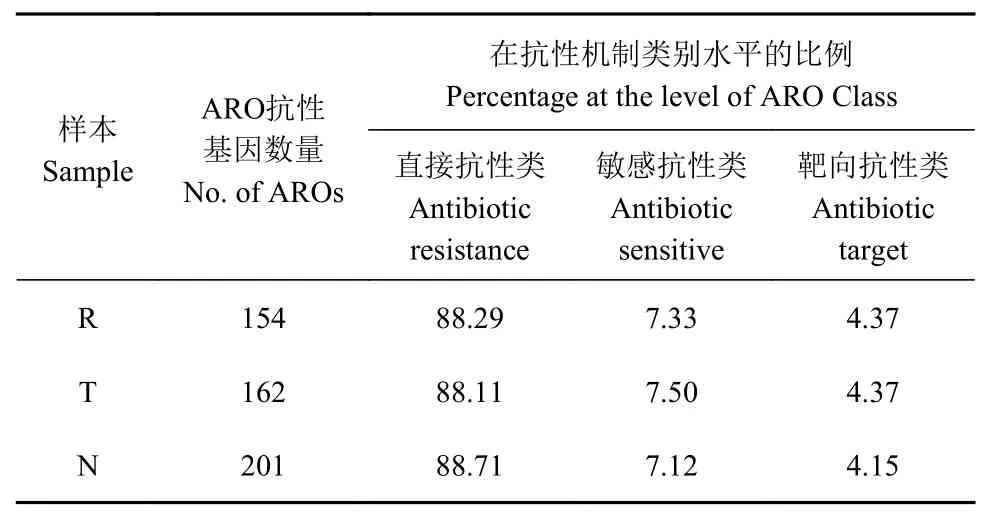
表6 不同样本中检测的ARO 抗生素抗性基因的数量及在抗性机制类别水平的比例Table 6 Amount and classified percentage of detected AROs in cultured samples (单位:%)
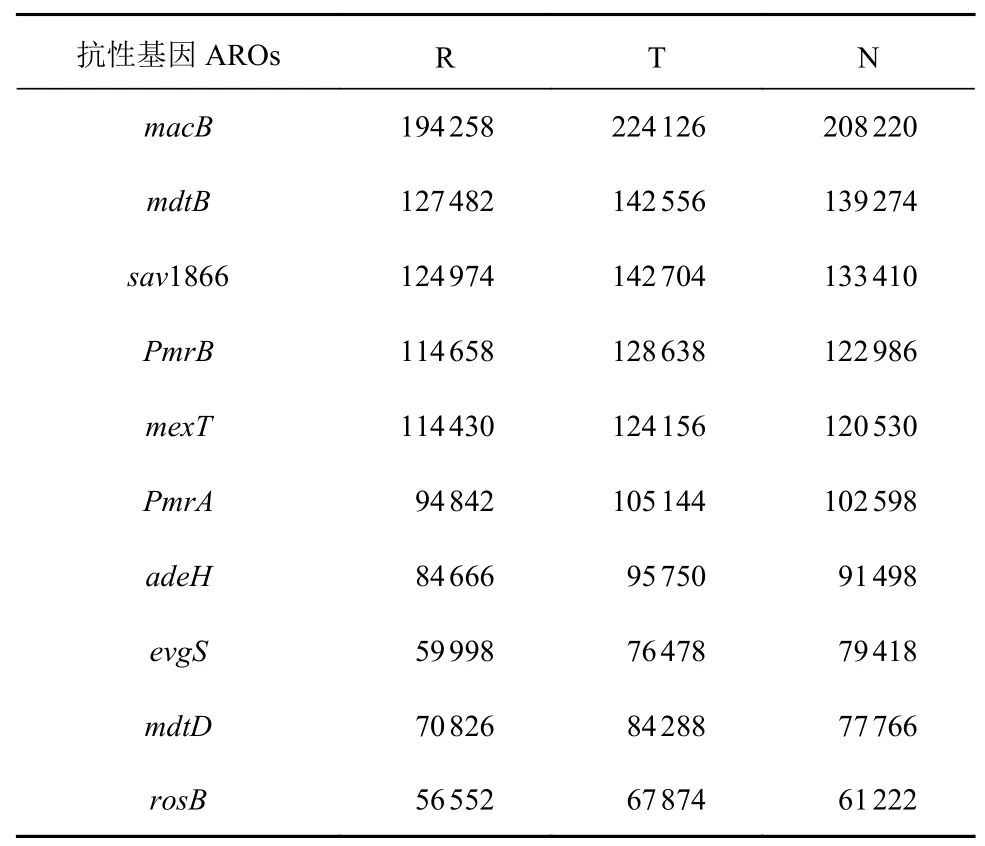
表7 不同样本中前10 位ARO 抗生素抗性基因的序列读长数Table 7 Read amounts of top 10 AROs in cultured samples
3 讨论与结论
本研究以培养无共生藻的宿主植物满江红的培养基Nickell[19]为对照,利用2 种通用的细菌培养基R2A,TRN 对小叶满江红的内生细菌进行了分离培养,发现3 种营养成分各异的培养组所培养形成的微生物物种基本是一致的。从门水平上而言,变形菌门(Proteobacteria)的细菌是其优势种群,推测这些种群的内生细菌很可能在满江红共生关系的营养代谢中起很大的作用。而通常在显微镜观测下占优势的蓝细菌门微生物在本培养中形成不了优势。从属水平而言,满江红内生细菌群落的优势度则更为的明显。结果证明,草螺菌属(Herbspirillum)可占到全部细菌菌属的90%以上,说明它是满江红内生菌中的一种优势菌种。已知草螺菌属(Herbaspirillum)是一种常见的植物内生菌,多分布于根茎部,且多具合成植物生长素、联合固氮等植物促生功能[23]。在富营养的培养基中成为满江红一种优势生长的内生菌,其机制值得研究。
近年来宏基因组分析技术在抗生素抗性基因的发现、传播机制、进化分析等方面均起到了至关重要的作用。这得益于两个方面:一方面,由于高通量测序技术的发展,整合工作流程至产生结果所花费的时间大大减少,能够在前所未有的短时间内对大群体的微生物进行比较,通过基因组测序,科研工作者可以快速了解目标微生物携带的耐药基因和可移动元件等[24];另一方面是由于抗性基因的生物信息学分析手段的进步,各类基因分析工具的开发和抗生素抗性基因数据库的建立[25]。ARDB 是一个人工搜索和校对的方法建立起来的微生物耐药基因数据库[21]。许多研究采用了ARDB 鉴定抗生素抗性基因[26-27]。本研究通过ARDB 比对,可以检测到小叶满江红内生细菌的特征耐药基因是baca与ceob。而CARD 是一个基于志愿者贡献数据的共享平台[22],每月更新一次以确保数据的时效性。本研究通过CARD 比对,注释了212 抗生素相关基因ARO。根据耐药机理[28-29],CARD 将每个ARO 以:产生灭活酶或钝化酶(antibiotic inactivation enzyme)破坏抗生素的活性;改变细胞壁或膜的渗透作用(altering cell wall charge)进而阻碍抗菌药物的进入;加快细菌主动外排作用(efflux pump),排出菌体内的抗生素;改变抗生素作用靶位(antibiotic target modifying enzyme)等方面进行具体功能描述,但在类别(Class)水平主要分为直接抵抗AR,敏感抵抗AS 和靶点结合AT 等类别。本研究发现的满江红内生细菌抗生素相关基因ARO包含了AR,AS 和AT 等3 个类别,且绝大多数是直接作用的AR 类抗性基因。
本研究通过对小叶满江红内生细菌的培养,首次结合宏基因组测序分析,在3 种不同营养成分的培养基中注释到了38 门712 属的物种。这说明宏基因组技术在对满江红内生微生物物种基因检测的广度和精度方面与前人比都有极大的提升[12-16],因为只要能检测到某物种的1 条序列读长(read),就可以加以注释。但考虑到基因丰度时,3 种样本均出现生长优势的特定微生物群落—变形菌门草螺菌属。鉴于本研究的宿主材料小叶满江红的来源单一,又长期在一种环境下进行种质资源保护式的繁殖生长,使得其内生细菌保持较纯的原生状态。推测草螺菌本是小叶满江红的内生的原生态菌之一,且适应性及繁殖能力极强,在3 种培养基中均含氮、碳等营养成分时,就可迅速繁殖成优势菌。而其他细菌,如易在显微镜下观察到的蓝细菌门的细菌虽然体型较大,但在培养基营养丰富时,竞争不过草螺菌而成为劣势菌群。对其中抗生素抗性基因的检测可以进一步推测:正是由于这些单一优势菌群的出现,仅能检测到满江红可携带为数不多的特定的抗生素抗性基因。本研究建立了水生植物满江红内生细菌的培养程序,并应用宏基因组技术对细菌的群落组成和所携带的抗生素抗性基因进行检测,为今后利用满江红对淡水环境中的抗生素抗性基因的动态状况进行监测提供了方法。通过对满江红内生微生物的特异基因的检测,也为筛查ARGs 在环境中依靠水生植物内生菌的扩散提供了新的思路。
猜你喜欢
江苏安全生产(2022年2期)2023-01-16 04:09:14
空间科学学报(2021年1期)2021-05-22 01:36:34
湖北农机化(2020年4期)2020-07-24 09:07:16
世界农药(2019年4期)2019-12-30 06:25:10
今日农业(2019年11期)2019-08-15 00:56:32
乡村地理(2018年2期)2018-09-19 06:44:02
环境保护与循环经济(2017年5期)2018-01-22 02:56:44
新农业(2017年2期)2017-02-02 11:10:59
新农业(2016年16期)2016-08-16 03:42:22
新农业(2016年14期)2016-08-16 03:33:15
