
氢化MoS2/石墨烯异质结的第一性原理计算
2021-08-17 08:10:34张国英张安国孟春雪
沈阳师范大学学报(自然科学版) 2021年3期
张国英,张安国,孟春雪
(沈阳师范大学 物理科学与技术学院,沈阳 110034)
0 引 言
十几年来,石墨烯因其优异的电学、机械、光学等性质而受到广泛关注,尤其是单层石墨烯电子迁移率极高,因而在微电子领域有着极大的潜力[1]。二硫化钼(MoS2)是二维层状过渡金属硫属化物(TMDCs)中的代表,块体MoS2为间接带隙,单层MoS2为直接带隙,是用来制作光探测器的优秀材料[2-5]。单层MoS2具有与石墨烯相似的结构,且可以晶格适配。由于石墨烯具有高电子迁移率和MoS2的光催化活性位点丰富的特点,MoS2/Gr异质结复合材料成为研究热点之一[6-8]。危阳等[9]用第一性原理计算发现,将石墨烯和MoS2制成异质结后带隙为1 meV;张秀云等[10]用第一性原理计算得到在MoS2/Gr异质结中加入过渡金属原子可以改变其能带;张琪瑛[11]发现,对石墨烯表面进行氢化后可以有效地改变石墨烯的能带结构,且加入的氢原子越多,带隙越宽。因此,本文采用第一性原理研究MoS2/Gr异质结及其氢化后的结构稳定性、电子性质和光学性质。本文通过对各个结构的结合能、体系结构、能带结构、电子态密度及光吸收系数进行分析,研究结构稳定性、电子性质及光学性质。
1 计算模型和方法
本文采用的是基于密度泛函理论的第一性原理MS(Material Studio)软件中的CASTEP完成计算。选择广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)[12],并利用Grimme的DFT-D方法来修正层间的范德瓦尔斯力[13]。在描述离子实与价电子之间的相互作用时,选取的价电子组态分别为C:2s22p2,S:3s23p4,Mo:4s24p64d55s1,布里渊区k点网格均选取为2×2×1,平面波截断能设置为350 eV,对晶胞内所有原子进行了完全弛豫,弛豫收敛精度为2.0×10-5eV/atom,原子间的力场收敛精度设置为0.5 eV/nm,最大应力设置为0.1 GPa,最大位移不超过2×10-4nm。

(a)正视图;(b)顶视图图1 层状MoS2Fig.1 Layered MoS2
采用GGA-PBE方法对六方相MoS2(空间群P63/mmc)的单胞进行几何结构优化,晶格常数为a=b=3.17 Å,c=12.32 Å;再对石墨烯的单胞同样进行几何结构优化,晶格常数为a=b=2.46 Å。结果与实验值的误差均小于1%(实验值:二硫化钼a=b=3.16 Å,c=12.30 Å;石墨烯a=b=2.46 Å)[14]。MoS2具有层状堆叠结构,从中剥离出层状MoS2,属于六角密堆结构,中间一层为Mo原子,上下两层均为S原子,形成S-Mo-S的三明治结构(图1)。为了使层状MoS2与石墨烯的晶格匹配,制作MoS2/Gr异质结时采用的模型为3*3周期结构的层状MoS2超晶胞和4*4周期结构的石墨烯超晶胞匹配(图2(a)),为了避免周期性相互作用,真空层厚度设定为20 Å。再在异质结表面加3个H(图2(b)),5个H(图2(c)),8个H(图2(d)),上下加3个H,5个H,8个H(图2(e)),上面加8个H下面加5个H(图2(f))。

(a)MoS2/Gr异质结顶视图;(b)MoS2/Gr+3H;(c)MoS2/Gr+5H;(d)MoS2/Gr+8H;(e)MoS2/Gr+8H+8H;(f)MoS2/Gr+8H+5H图2 各体系结构图Fig.2 Diagrams of various architectures
2 结果及讨论
2.1 结构稳定性
为了得到MoS2/Gr异质结的稳定结构,石墨烯中的C原子固定在MoS2中六边形的中心位置进行结构弛豫,得到图2(a)的稳定结构模型。经过弛豫后,异质结的层间距为3.39 Å,比石墨烯的层间距(4.01 Å)更短,说明MoS2与石墨烯为较弱的范德华力相结合。与复合前的单组份相比,复合后的结构几乎没有变化。MoS2及石墨烯的超晶胞的晶格常数分别为a1,a2,晶格失配率为a=(a1-a2)/a1。由此得到MoS2/Gr的晶格失配率大约为3.7%,小于5%,可视其为完全共格。
为了进一步讨论结构稳定性,计算了其结合能。MoS2/Gr异质结体系的所有原子与晶胞均被充分弛豫优化,异质结的结合能与对异质结氢化时的氢原子结合能分别为
Eb=Et-EGr-EMoS2
其中:Et,EGr,EMoS2分别为异质结体系的总能量、石墨烯和单层MoS2的能量;EH表示1个H原子的能量;n表示加入H原子的个数;Eb为负值,其绝对值越大,表示结构越稳定。
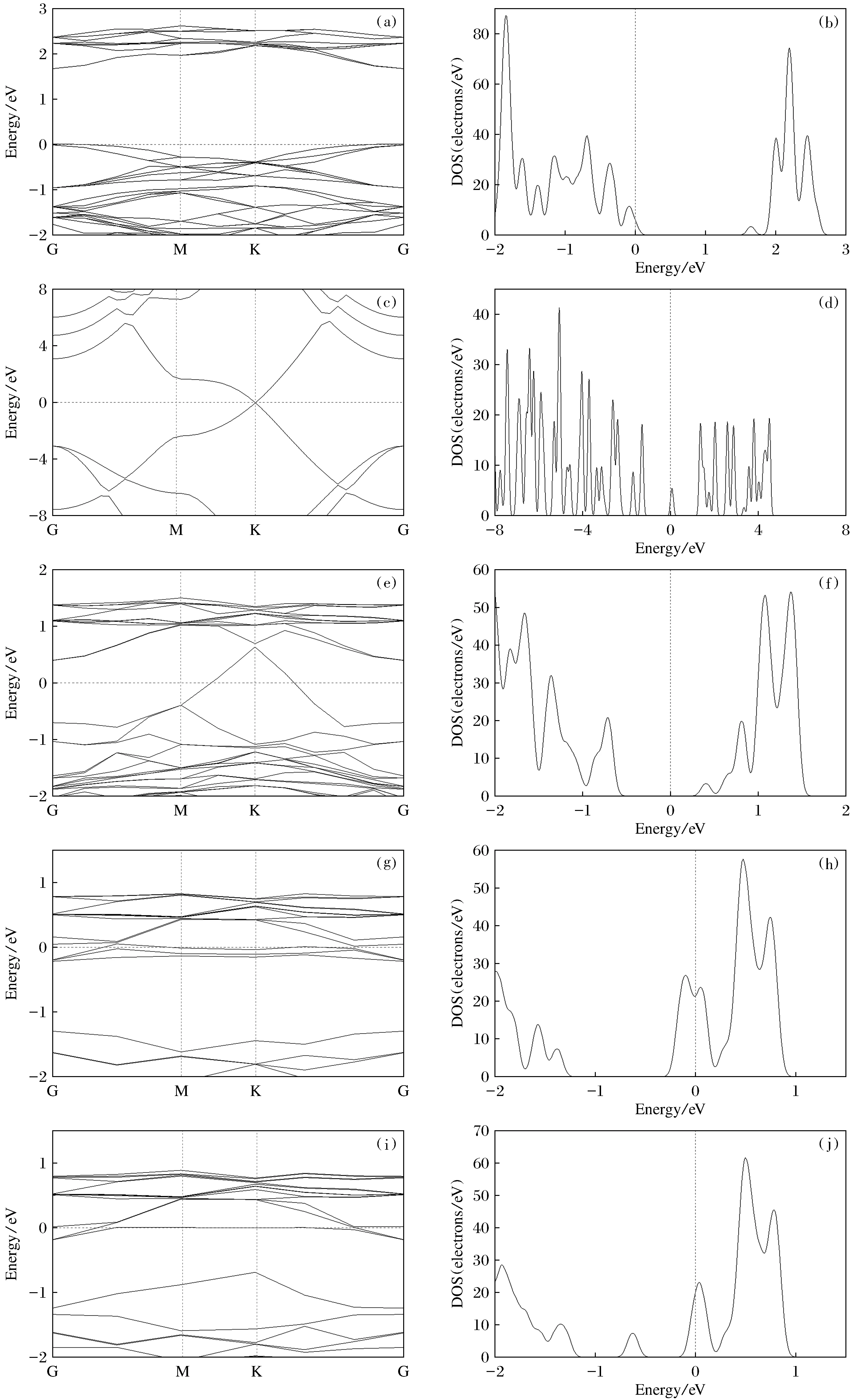
经过计算,MoS2/Gr异质结及氢化后的结合能Eb/eV,MoS2与石墨烯的层间距D/Å,C原子与其表面H原子的最小间距LC—H/Å、MoS2中的S原子与其表面H原子最小间距LS—H/Å如表1所示。由表1可知,在MoS2/Gr异质结及其表面进行氢化后的8种结构的结合能都为负数,其结构是稳定的。MoS2/Gr异质结的层间距为3.39 Å,在石墨烯上表面氢化后,层间距会加大10%左右,不同体系的层间距与加入的H原子个数有关,且加入的H原子越多,层间距越大。在MoS2/Gr异质结的上下表面分别氢化后,体系的层间距会加大,同样遵循加入的H原子个数越多,体系的层间距越大的特点。但与只在上表面进行氢化相比,在上下表面同时进行氢化的体系层间距会相应地小一些(D3H 表1 MoS2/Gr异质结及氢化后的结合能Eb/eV,层间距D/Å,C—H间距LC—H/Å,S—H间距LS—H/ÅTable 1 MoS2/Gr heterojunction and its hydrogenated binding energy Eb/eV,interlayer spacing D/Å,C-H spacing LC-H/Å,S-H spacing LS-H/Å 为了研究MoS2/Gr异质结及对异质结表面进行氢化后的界面结构和电子性质,计算了前面提到的所有结构的能带结构及态密度。图3为单层MoS2,石墨烯和MoS2/Gr异质结及氢化后异质结的能带结构及态密度。可以看出,单层MoS2显示出高对称G点位置的直接带隙,禁带宽度为1.648 eV(图3(a)),实验结果为1.8 eV。可以发现理论计算值略低于实验值,这是因为密度泛函理论在计算带隙时普遍存在不可避免的误差,GGA近似往往会高估晶格常数并低估带隙,但这并不影响带隙结构及其他方面结果的分析。石墨烯表现为零带隙(图3(c)),价带顶和导带底均在K点。当将MoS2与石墨烯制成异质结后,禁带宽度变成了0.055 eV(图3(e))。与石墨烯相比,异质结的费米能级发生了移动,位于石墨烯狄拉克点的下方,费米能级位于MoS2的导带底附近。并且石墨烯狄拉克点的位置高于单层MoS2价带底的位置,即存在电势差。对异质结的石墨烯上表面进行氢化,MoS2/Gr+3H和MoS2/Gr+5H的带隙均为零(图3(g)和(i))。MoS2/Gr+8H的禁带宽度为1.076 eV(图3(k)),价带顶和导带底均处于G点,直接带隙表现为半导体,与Si的带隙(1.1 eV)较为相似。在对石墨烯上表面及MoS2下表面进行氢化后,MoS2/Gr+3H+3H和MoS2/Gr+5H+5H表现为零带隙。MoS2/Gr+8H+8H的禁带宽度为0.475 eV(图3(m)),并且出现在高对称G点位置的直接带隙,与MoS2/Gr异质结相比明显加宽了异质结的带隙。但是有3个H原子逃逸到晶胞之外,故计算了MoS2/Gr+8H+5H,禁带宽度为0.220 eV(图3(s))。由此不难看出,在MoS2/Gr异质结表面进行氢化是可以调节体系的带隙的。为了更好地了解MoS2/Gr异质结及对异质结进行氢化后的层间相互作用的微观机理,计算了各体系的总电子态密度(图3(b),(d),(f),(h),(j),(l),(n),(p))。从态密度图中可以得到同样的结论。 (a),(b)MoS2;(c),(d)石墨烯;(e),(f)MoS2/Gr异质结;(g),(h)MoS2/Gr+3H;(i),(j)MoS2/Gr+5H;(k),(l)MoS2/Gr+8H;(m),(n)MoS2/Gr+8H+8H;(o),(p)MoS2/Gr+8H+5H图3 不同结构的能带结构和态密度图Fig.3 Band structure and density of states diagrams of different structures 材料的基本光学性质有折射率、消光系数、反射率和吸收系数,只需要知道这4种基本光学常数就可以描述和表征材料的所有光学性质,并且这4个光学常数都可以由介电常数ε(ω)表示[15]。ε(ω)=ε1(ω)+iε2(ω),其中ω为吸收光的频率,介电常数的实部与虚部均可通过密度泛函理论计算得出。介电常数的虚部对应于电子跃迁的几率和强度。为了讨论MoS2/Gr异质结及在其表面进行氢化后的各体系的光学性质,对各个体系光学跃迁特性变化进行研究。通过计算其介电常数,得出它们的光吸收谱。通过光吸收谱可以清晰地看出各体系光吸收系数与光子波长之间的关系,其光吸收系数的公式如下: 计算发现,MoS2/Gr异质结及其在表面氢化后的结构的光吸收峰均出现在220 nm左右,且在200~280 nm的光吸收率明显高于其他区域,为紫外光区。在只对上表面进行氢化的几种结构体系中,220 nm的光吸收率随着附氢的数量的增加而增加,具体表现为α(8H)>α(5H)>α(3H)。在可见光区(380~780 nm),氢化程度越高,吸收率越高,具体表现为α(8H)>α(5H)>α(3H)>MoS2/Gr,α(8H+8H)>α(8H+5H)>α(5H+5H)>α(3H+3H)。 本文基于第一性原理计算的密度泛函理论,研究了氢化对MoS2/Gr异质结的调控机制。分析了在MoS2/Gr异质结石墨烯上表面和MoS2下表面添加不同个数的氢原子的结构稳定性、能带结构、总态密度及光学性质。结果表明在MoS2/Gr异质结表面进行氢化后的结构都是稳定的。MoS2/Gr异质结本身的带隙较小,在对其石墨烯上表面或在石墨烯上表面与MoS2下表面分别氢化后,异质结的能带会被打开,且加入的氢原子越多,带隙越大。尤其是在石墨烯上表面加入8个氢原子后能带变成了1.076 eV,表现为半导体。可见对MoS2/Gr异质结体系进行氢化可以改变异质结的带隙。这对类石墨烯材料的能带调控提供了积极意义。将二硫化钼与石墨烯制成异质结后,光吸收峰出现在220 nm的紫外光区。在对异质结表面进行氢化后,光吸收峰的位置并未发生变化,但在可见光区的光吸收率增加了,且加入的氢原子越多,在可见光区的光吸收率越高。这对二硫化钼/石墨烯异质结材料在利用可见光方面提供了可能,并为其在光电方面的应用提供了积极意义。
2.2 电子性质

2.3 光学性质
3 结 论
猜你喜欢
小天使·聪聪画刊(2021年2期)2021-09-10 07:22:44
汽车零部件(2020年10期)2020-11-09 03:41:42
汉语世界(The World of Chinese)(2019年6期)2019-09-10 07:22:44
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:16
橡胶工业(2015年2期)2015-07-29 08:29:46
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:26
物理实验(2015年10期)2015-02-28 17:36:52
西南军医(2014年5期)2014-04-25 07:42:49
食品工业科技(2014年9期)2014-03-11 18:15:39
无机化学学报(2014年5期)2014-02-28 17:31:39
