
基于分子动力学模拟的Vmo Lac非特异性底物催化活性的理论研究
2021-08-16 05:14:26李聪聪刘明皓韩佳睿朱镜璇韩葳葳李婉南
高等学校化学学报 2021年8期
李聪聪,刘明皓,韩佳睿,朱镜璇,韩葳葳,李婉南
(吉林大学分子酶学工程教育部重点实验室,生命科学学院,长春 130012)
有机磷农药在我国的生产和使用广泛,但其使用后会不同程度地残留在农作物中,造成严重的土壤和水污染,破坏生态平衡,并且对生物体产生直接或间接的毒性,极大地威胁人类健康[1].现有的去除有机磷类残留物方法产生的负面效果较多,如成本高,反应剧烈,存在安全隐患等,因而难以得到广泛应用.细菌磷酸三酯酶(PTEs)是目前存在的能有效降解有机磷农药残留的一类酶[2~4].常温下,磷酸三酯酶稳定性较差,而嗜热磷酸三酯酶样内酯酶(PLLs)是具有嗜热性的一类天然内酯酶,在高温下具有高反应活性,因此PLLs可作为残留有机磷农药降解的靶标蛋白[5,6].PLLs按照结构特点和催化活性可分为PLLs-A(天然的酰基高丝氨酸内酯酶)家族和PLLs-B(氧合内酯酶)家族[7,8].
VmoLac是来自极端嗜性古细菌V.Moutnovskia(YP_004245953)的PLLs-A家族成员[9],其可在60~98℃下稳定存在[10,11],并在80℃时活性最高[12],比其超耐热同家族SsoPox的高出20℃以上.VmoLac与其它热稳定酶[13]具有许多共同的结构决定因素,如结构表面存在大量的盐桥以及较大的疏水性同源二聚体界面.VmoLac的活性位点Loop8呈刚性并且形成了α螺旋结构,与最接近的PLL(50%序列同一性)仍有很大不同,这种差异会对VmoLac结合口袋产生重要影响.2015年,Hiblot等[9]提出水解内酯酶的机理:VmoLac与其底物3-oxo-C10-AHL结合的结构能证明酶与结合的内酯具有特异性相互作用,VmoLac对内酯的水解机理与之前提出的SsoPox水解内酯的机理相似.内酯环结合到双金属中心上可使内酯中sp2杂化的碳原子更亲电,并释放出活化的桥接水分子.后者随后可通过由β-金属稳定的带负电荷的四面体中间体攻击内酯中sp2杂化的碳原子.同时内酯环上的羰基氧和Y98相互作用,含氧阴离子上的电子对折回,使酯键断裂,形成羧酸和醇化物.该醇化物可能需要酸性辅助,D257使剩下的醇化物质子化.因此,D257与极化水分子可以通过氢键连接起来,然后通过双金属中心与底物中内酯环的羰基碳原子、对氧磷的磷原子发生亲核反应而激活催化反应.
基于此,本文对VmoLac的非特异性底物催化活性进行了理论研究.采用分子对接和分子动力学模拟基于PLLs的催化水解机理,从蛋白质构象变化的角度探究了VmoLac非特异性底物的催化活性,并通过研究4种底物分别与VmoLac结合的能力和模式,比较了其催化水解长链内酯(3-oxo-C10-AHL)、短链内酯(3-oxo-C6-AHL)、壬内酯(γ-nonalacton)以及对氧磷(ethyl-paraoxon)的能力.为VmoLac非特异性底物催化活性的研究提供了重要的理论依据.
1 模拟方法
1.1 底物优化及分子对接
模拟所需的初始蛋白质晶体结构(PDB code:4RDY)[9]取自PDB数据库,4种底物3-oxo-C10-AHL,3-oxo-C6-AHL,γ-nonalacton和ethyl-paraoxon来自于Chemspider数据库.在B3LYP/6-31G*水平上优化4个底物的结构得到最优构象,并进行了密度泛函理论(DFT)计算以得到最高占据分子轨道-最底未占据分子轨道(HUMO-LUMO)带隙能量,计算均采用Gaussian 09[14]程序并用Multiwfn程序[15]对得到的分子轨道进行可视化分析.采用Autodock4.2[16,17]软件将小分子配体3-oxo-C10-AHL,3-oxo-C6-AHL,γ-nonalacton和ethyl-paraoxon使用半柔性对接方式对接到VmoLac的活性口袋中.根据已经确定VmoLac晶体结构的底物结合位置[9],将对接盒子的大小设为x=40,y=35,z=30,盒子的中心设为x=2.251,y=14.204,z=20.115,每个格点长度为0.0375 nm.采用拉马克遗传算法进行分子对接计算,从对接结果聚类最多的一类中选取能量最低的结构用做分子动力学模拟的初始结构.
1.2 分子动力学模拟
分别对VmoLac/3-oxo-C10-AHL,VmoLac/3-oxo-C6-AHL,VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon 4个复合物进行100 ns的分子动力学模拟,并对每个体系做3次平行模拟.所有分子动力学模拟均采用GROMACS程序[18,19]运行,使用AMBERff99SB力场[20]构建蛋白质的参数文件,小分子配体用GAFF力场[21]处理,其拓扑文件通过执行ACPYPE程序[22]生成.每个体系的蛋白质-配体复合物都使用TIP3P水模型[23]在尺寸为1 nm×1 nm×1 nm的周期性立方体盒子下进行MD模拟,为了使复合物保持电中性并确保离子浓度为150 mmol/L,向每个体系随机加入了适量的抗衡离子.每个体系的水分子和Na+数量如表1所示.

Table 1 Number of water and Na+for four simulation systems
采用最陡下降法使体系能量最小化.随后进行体系两个阶段的预平衡模拟,即100 ps的NVT模拟(Berendsen温度耦合恒定的颗粒数、体积和温度)和100 ps的NPT模拟(Parrinello Rahman压力耦合恒定的颗粒数、压力和温度).温度和压力的耦合常数分别设为0.1和2.0 ps,NPT模拟之后,盒子中的体系密度已达到最优状态,此时盒子的尺寸为0.85 nm×0.85 nm×0.85 nm.所有的键长和键角均采用P-LINCS[24]进行约束,并使用PME方法[24]处理长程静电相互作用.范德华相互作用的截断值为1.4 nm.待所有热力学性质稳定后,对4个体系分别进行步长为2 fs,每隔2 ps保存一次坐标文件,以便分析使用.
1.3 协方差分析
使用Bio3D[25,26]进行协方差分析,通过检测所有成对交叉相关系数的大小来评估系统的原子波动/位移与其它系统之间的相关程度[27].Bio3D的作用是生成一个由所有原子相互关系组成的矩阵,其元素可以用图形的形式显示出来,通常称为动态相互关系映射(Dynamic cross-correlation map,DCCM).在协方差图中,坐标轴比例尺对应于原子序数.红色区域颜色越深表示矩阵元素值越正,两种原子运动模式之间的相关性越正,与矩阵元素值一致的白色区域为0.蓝色区域越多,表示矩阵元素值越负,说明对应的两种原子运动模式趋于负相关.矩阵的对角线是方差,必须大于零,所以其呈白色或红色.
2 结果与讨论
2.1 底物结构优化及其反应位点预测
模拟3-oxo-C10-AHL,3-oxo-C6-AHL,γ-nonalacton和ethyl-paraoxon的三维(3D)结构如图1所示.

Fig.1 3D structures of 3-oxo-C10-AHL,3-oxo-C6-AHL,γ-nonalacton and ethyl-paraoxon
通过DFT计算后用Multiwfn程序分析得到4种底物的LUMO和HUMO轨道的能量值以及能量差如图2所示.图中给出了化合物由HOMO轨道跃迁至LUMO轨道的电子分布变化以及跃迁的能量差(Egap,表示电子跃迁所需能量的大小,即化合物发生反应趋势的大小).对比图2(A)和(B)可知,3-oxo-C10-AHL跃迁的能量差值(Egap)小于3-oxo-C6-AHL,说明前者更易发生电子转移,且两个化合物电子在轨道间跃迁的转移主要发生在碳链处,由此可推测内酯链的长短对反应的发生具有重要影响.由图2(C)和(D)可知,γ-nonalacton的LUMO轨道的电子集中分布在内酯环上,所以该环易发生亲核反应,而ethyl-paraoxon不存在内酯环,其亲核反应只能发生在磷原子上,且磷原子没有电子的分布,所以发生反应的趋势较弱.通过以上两个底物的比较可知,前者的Egap值小于后者,即壬内酯比对氧磷更易发生电子跃迁.综上,3-oxo-C10-AHL和γ-nonalacton更易参与亲核反应及与酶结合发生催化反应.

Fig.2 Electron orbital and orbital energy difference between HOMO and LUMO orbital for 3-oxo-C10-AHL(A),3-oxo-C6-AHL(B),γ-nonalacton(C)and ethyl-paraoxon(D)
2.2 分子对接及底物与Vmo Lac的相互作用
图3 (A)为VmoLac与3-oxo-C10-AHL复合物的3D结构图,图3(B)为VmoLac单体结构的表面静电势分布图,带负电荷的区域用蓝色表示,带正电荷的区域用红色表示,不带电荷的区域用绿色表示.活性位点口袋即底物结合部位用黄色矩形突出标记.结果表明,底物相对于晶体结构配体的位置偏移小于2 nm(图S1,见本文支持信息).4个复合物的结合自由能分别为-59,-46,-75和-54 kJ/mol.

Fig.3 Overview of Vmo Lac/3-oxo-C10-AHL(PDBcode:4RDY)(A)and the surface electrostatic potential of Vmo Lac(B)
将对接得到的复合物VmoLac/3-oxo-C10-AHL,VmoLac/3-oxo-C6-AHL,VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon通过PLIP服务器进行配体与VmoLac之间的相互作用分析[28].4种底物与蛋白的相互作用结果如图4所示,Loop8(Y356—T280)是位于VmoLac的活性中心附近且对酶发生催化反应具有重要影响的区域,由图4(A)可知,Loop8区域中可以与3-oxo-C10-AHL的碳链部分发生疏水相互作用的残基包括Y265,V269,T273,V274和W277,而与3-oxo-C6-AHL产生疏水作用的残基仅包括Y265,V274和W277,可见,长链内酯更易与酶的Loop8区域发生疏水作用,进而保证长链可以有足够的空间进入活性中心,短链内酯却相反.由图4(C)和(D)可见,γ-nonalacton和ethyl-paraoxon与酶的相互作用差异较大,与对氧磷作用的残基有L28,W224,I229,Y230,I262,W264,Y265和W277.其中Y265与其产生共轭相互作用.对氧磷底物与内酯底物不同的是对氧磷中不含内酯环,因此不存在Y98与对氧磷的相互作用.Y98是影响VmoLac特异性催化底物的重要残基,并影响VmoLac催化Paraoxon的活性.

Fig.4 Interaction diagram between 3-oxo-C10-AHL(A),3-oxo-C6-AHL(B),γ-nonalacton(C),ethyl-paraoxon(D)and Vmo Lac generated with PLIP
2.3 分子动力学模拟过程中蛋白质结构的稳定性
分析了4个体系在分子动力学模拟过程中不同配体结合对蛋白质结构稳定性影响的差异.蛋白质Cα原子骨架的均方根偏差(RMSD)可用于表征蛋白质构象差异程度,回转半径(Rg)可以反映蛋白质结构紧密程度.4个复合物动力学模拟的RMSD结果及标准差如图5所示,可见,重复实验的误差值在合理范围内,模拟体系达到平衡,可以保证结论的可靠性.
根据每个体系中蛋白质的RMSD和Rg值的概率密度分布绘制的自由能分布如图6所示,自由能值越低分布概率越大.由图6(A)可见,VmoLac/3-oxo-C10-AHL体系的主能量盆位于RMSD值(~0.175nm)和Rg值(~1.915 nm)之间,而对于VmoLac/3-oxo-C6-AHL复合物,主能量盆在RMSD值(~0.275 nm)和Rg值(~1.835 nm)之间[图6(B)],

Fig.5 Root-mean-square deviation(RMSD)and standard deviation plot of the repeated simulations for Vmo Lac/3-oxo-C10-AHL(A),Vmo Lac/3-oxo-C6-AHL(B),Vmo Lac/γ-nonalacton(C)and Vmo Lac/ethyl-paraoxon(D)
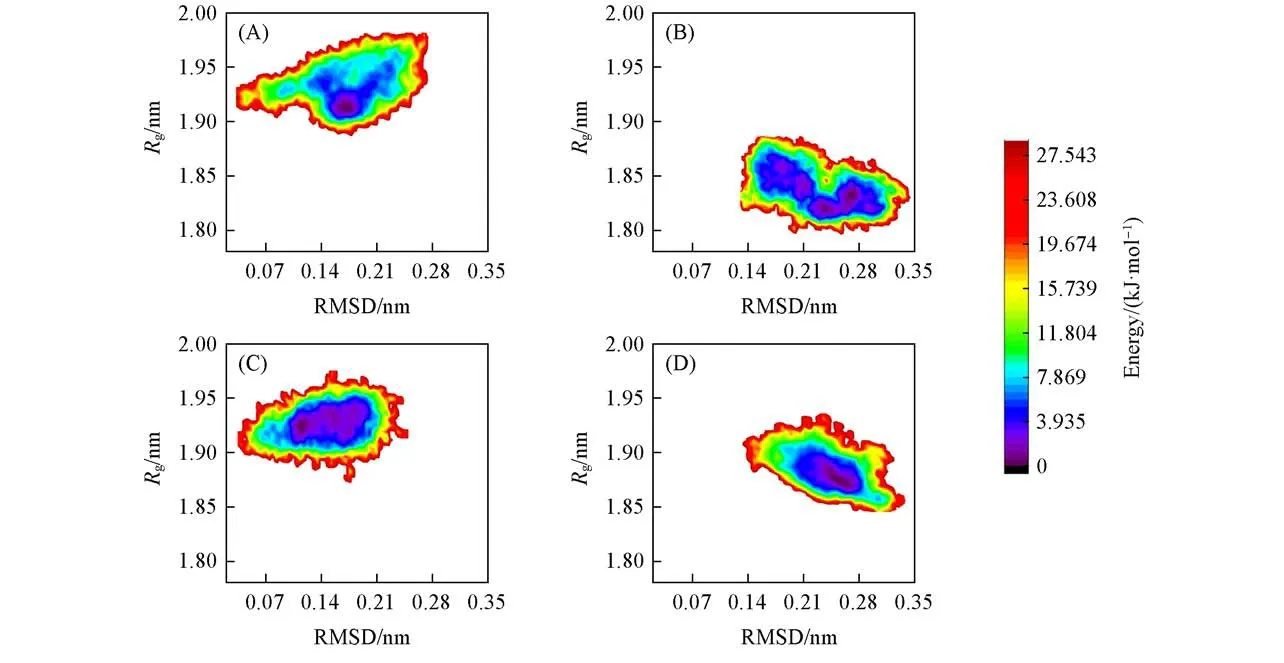
Fig.6 Free energy landscape for R g and RMSD of Vmo Lac/3-oxo-C10-AHL(A),Vmo Lac/3-oxo-C6-AHL(B),Vmo Lac/γ-nonalacton(C)and Vmo Lac/ethyl-paraoxon(D)
可以看出后者的RMSD值明显高于前者,而Rg值低于前者.两者RSMD值的差异说明3-oxo-C10-AHL结合会使VmoLac更稳定,Rg结果的差异表明当底物是3-oxo-C10-AHL时,VmoLac的蓬松程度更大,更有利于底物进入酶的活性口袋与之结合.图6(C)和(D)分别为VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon的自由能分布图,分析原理与前两个体系相同.结果表明,γ-nonalacton比paraoxon与VmoLac结合更稳定,VmoLac/γ-nonalacton体系的蛋白在运动过程中更蓬松,更易于底物与酶的结合并发生催化作用.
均方根波动(RMSF)可用于计算单个残基的波动幅度,并用于衡量在动力学模拟过程中原子运动的自由程度,从而可以反映蛋白质区域运动的灵活性.4个体系在平衡状态下计算RMSF得到的结果如图7所示,Loop8(Y265—T280)区域的RMSF值在不同体系中表现出明显的差异性.
由图7(A)可见,VmoLac/3-oxo-C10-AHL比VmoLac/3-oxo-C6-AHL复合物中Loop8结构域中残基的运动自由度高,由图7(B)可见,与VmoLac/ethyl-paraoxon体系相比,VmoLac/γ-nonalacton体系的Loop8区域的RMSF值明显大得多,说明该区域的残基运动自由度高.Loop8区域位于VmoLac酶的活性区域附近,对酶的催化活性有重要影响,所以Loop8区域的运动自由度高会使3-oxo-C10-AHL和γ-nonalacton更易进入酶的活性口袋区域,并且更好地结合在疏水通道上,促进VmoLac对长链内酯和壬内酯的催化水解作用.
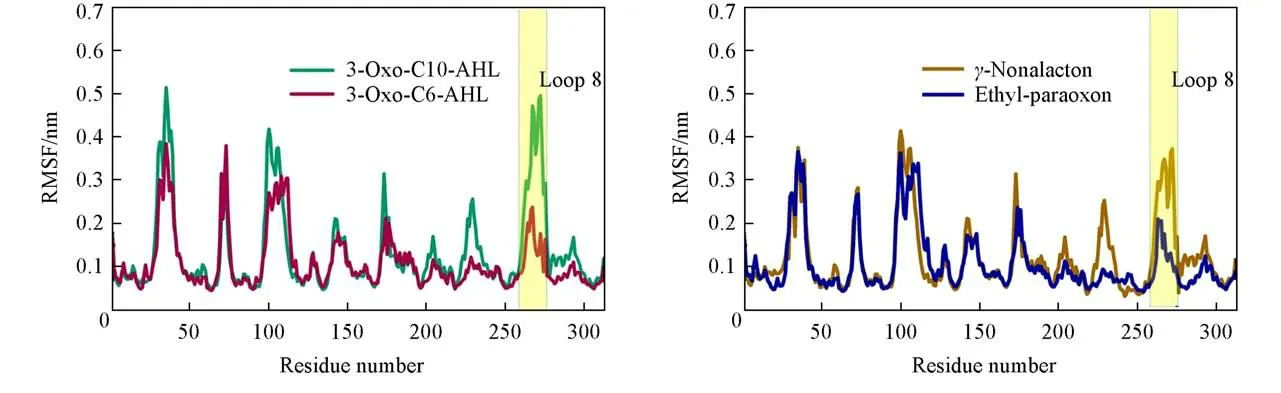
Fig.7 Comparison of the RMSF plots of proteins in Vmo Lac/3-oxo-C10-AHL and Vmo Lac/3-oxo-C6-AHL(A)Vmo Lac/γ-nonalacton and Vmo Lac/ethyl-paraoxon(B)complexs
2.4 配体结合对蛋白质结构运动的影响
为了从构象变化的角度解释蛋白质与配体之间的相互作用以及蛋白质残基之间的运动相互联系,对分子间的运动相关性进行了分析,即协方差分析.图8给出了蛋白质区域强烈的相关性构象变化.通常,代表正值的红色区域表示残基的强烈相关运动,代表负值的蓝色区域可能与负相关运动有关.对角线区域显示出最强烈的正相关运动,因为其是残基与自身的相关运动.由图8(A)和(B)可见,通过对比VmoLac/3-oxo-C10-AHL体系和VmoLac/3-oxo-C6-AHL体系中Loop8区域的残基运动相关性可知,前者明显比后者在Loop8区域的正相关性强并且在动力学模拟过程中波动幅度较大,由于Loop8区域位于VmoLac的活性中心附近,Loop8的运动有利于帮助3-oxo-C10-AHL的长链进入活性口袋发生催化反应.而与3-oxo-C6-AHL结合的Loop8区域因运动幅度较小而不利于短链内酯进入活性口袋,因此长链内酯比短链内酯更易结合于VmoLac的活性口袋区域而发生催化反应.
VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon复合物中底物结合对蛋白质残基之间相互影响的分析如图8(C)和(D)所示.与前者相比,后者的Loop8区域在互相关系矩阵图中表现为正相关性较低,在动力学模拟过程中Loop8区域运动不明显.分析原理与上两个复合物相同,表明壬内酯比对氧磷更易与VmoLac的活性口袋区域结合而促进催化反应发生.与结构稳定性分析的结果一致.

Fig.8 Dynamical cross-correlation map for the 100 ns MD simulation trajectories of the Vmo Lac/3-oxo-C10-AHL(A),Vmo Lac/3-oxo-C6-AHL(B),Vmo Lac/γ-nonalacton(C)and Vmo Lac/ethyl-paraoxon(D)complexes
2.5 Vmo Lac对非特异性底物催化活性的理论分析
VmoLac中的W264是底物结合口袋附近的重要残基,当活性中心的金属离子使溶剂的水分子去质子化时,会生成氢氧根离子,这时W264能与生成的氢氧根离子以及β金属离子发生桥连而产生相互作用,有利于氢氧根离子的亲核进攻.Y230是影响底物与VmoLac结合的重要残基.W264和Y230都位于酶的活性口袋中心的入口附近,可认为是底物进入酶的活性口袋的门控残基,因此在分子动力学模拟过程中这两个重要残基的构象重构或者位置重排都将会引起它们之间的距离变化,此变化会改变酶的活性中心口袋的入口处的大小,从而使底物进出活性中心口袋受到影响,最终影响非特异性底物的催化活性.因此,对4个模拟体系VmoLac/3-oxo-C10-AHL,VmoLac/3-oxo-C6-AHL,VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon的轨迹计算了W264和Y230中Cα原子之间的距离随模拟时间的变化.图9(A)展示了W264和Y230以及底物位置的相对位置示意图,两个残基中的Cα原子在动力学模拟过程中的距离变化情况如图9(B)和(C)所示,为了保证结果的可靠性,对4个体系3次平行模拟的W264与Y230之间的距离进行重复计算,结果如图S2(本文支持信息)所示.在VmoLac/3-oxo-C10-AHL中,W264和Y230的Cα原子距离保持在2.1 nm左右,而对于VmoLac/3-oxo-C6-AHL,此距离明显小于2.1 nm;VmoLac/γ-nonalacton体系的Cα原子距离明显大于VmoLac/ethyl-paraoxon体系.两个残基之间的距离小会对拟进入活性中心口袋的底物产生空间位阻,不利于底物进入活性中心口袋,进而影响酶的催化活性.所以VmoLac对长链内酯(3-oxo-C10-AHL)催化活性强于短链内酯(3-oxo-C6-AHL).同理,γ-nonalacton比ethyl-paraoxon更容易进入VmoLac活性中心口袋发生催化作用.
PLLs水解内酯以及对氧磷的机理表明,在内酯酶中,Y98可与底物内酯中的羰基碳或者对氧磷中的磷原子形成较强的氢键相互作用,从而使碳原子表现出更强的亲电性质.所以,Y98与内酯环中的羰基碳或对氧磷中的磷原子之间的距离被定义为底物的进攻距离.为了研究VmoLac与4种不同底物结合时进攻距离在分子动力学模拟过程中的变化,分析了4个体系在100 ns模拟时长内Y98与内酯环中的羰基碳(C=O)或者对氧磷中磷原子中心(P=O)之间距离的变化.图10(A)和(B)分别给出了Y98与底物亲电中心原子中间的3D结构及相对位置,二者在不同体系中的进攻距离变化如图10(C)和(D)所示,Y98与长链内酯3-oxo-C10-AHL的羰基碳之间的距离稳定在0.25 nm附近,而与短链内酯3-oxo-C6-AHL之间的距离约在0.28 nm,明显大于前者,表明在与VmoLac发生催化作用时3-oxo-C10-AHL的进攻距离比3-oxo-C6-AHL的小,催化反应更易发生.在VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon体系中,通过比较两个体系Y98与底物亲电中心原子(内酯环的羰基碳或对氧磷中的磷原子)之间距离可知,VmoLac/ethyl-paraoxon复合物的进攻距离较大,所以VmoLac催化对氧磷的活性较弱,而内酯的进攻距离较小.综上,内酯比对氧磷更适合作为VmoLac的底物,同时长链内酯比短链内酯更适合与VmoLac发生催化反应.
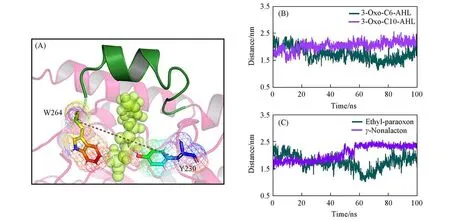
Fig.9 3D structure of residues W264 and Y230(A)and distance between the Cαof W264 and Y230 for Vmo Lac/3-oxo-C10-AHL and Vmo Lac/3-oxo-C6-AHL(B),Vmo Lac/γ-nonalacton and Vmo Lac/ethyl-paraoxon(C)during 100 ns MD simulations
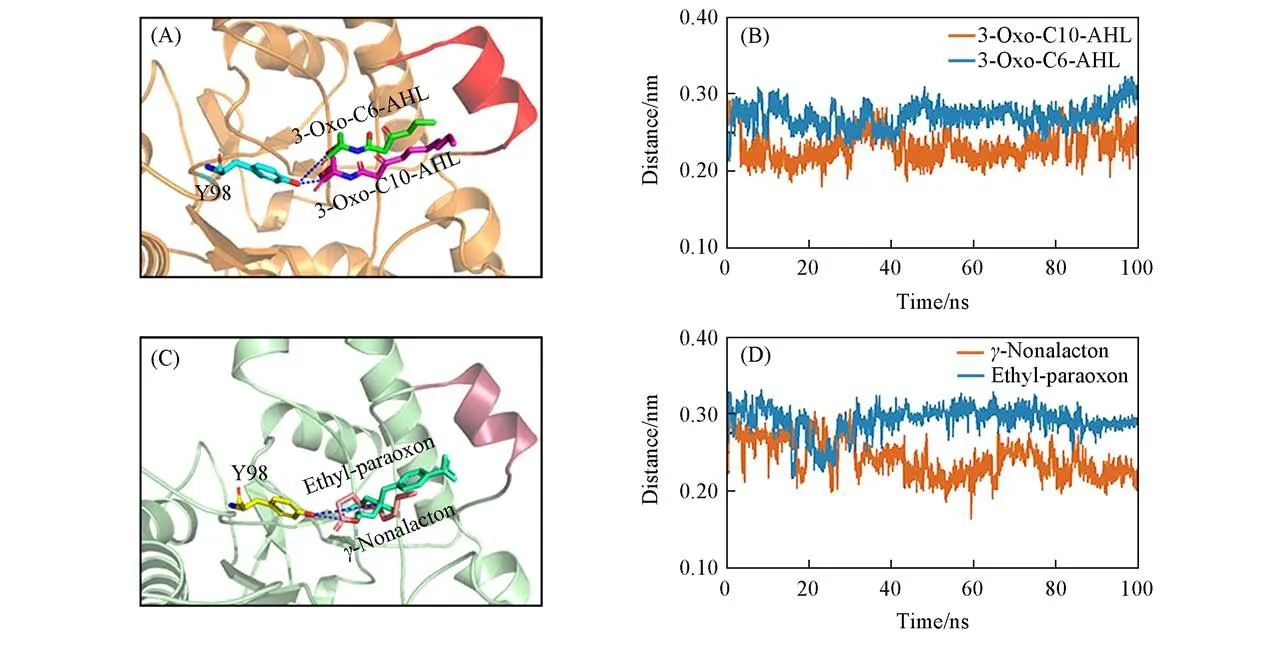
Fig.10 3D structures of Y98 and 3-oxo-C10-AHL,3-oxo-C6-AHL(A),the position of Y98 andγ-nonalacton,ethyl-paraoxon(B),the distance between the Y98 and electrophilic central atom of the substrate for Vmo Lac/3-oxo-C10-AHL and Vmo Lac/3-oxo-C6-AHL(C),Vmo Lac/γ-nonalacton and Vmo Lac/ethyl-paraoxon(D)during 100 ns MD simulations
VmoLac中的残基D257可与极化的水分子形成氢键,通过双金属离子的桥连作用对内酯环中的羰基碳和对氧磷中的磷原子发生亲核进攻反应,从而促进催化反应的激活.为了研究4个复合物在分子动力学模拟过程中底物与VmoLac之间结合能力的差异,用VMD1.9.1的氢键分析插件计算了在100 ns时长的动力学模拟期间D257与极化水分子以及不同底物之间氢键的形成几率.VmoLac/3-oxo-C10-AHL和VmoLac/3-oxo-C6-AHL体系中氢键形成几率列于表2,可以看到,在VmoLac/3-oxo-C10-AHL体系中,D257与极化水之间形成的氢键几率大于VmoLac/3-oxo-C6-AHL体系.其中,前者D257中的O原子与水分子中的O原子形成氢键的几率为83.21%,后者则仅为69.25%.说明相对于VmoLac/3-oxo-C6-AHL,在VmoLac/3-oxo-C10-AHL复合物中,D257可以与水分子形成了更强的氢键,即对3-oxo-C10-AHL的羰基碳具有更强的亲核进攻作用,所以VmoLac水解3-oxo-C10-AHL的能力更强.通过比较两种复合物中D257与底物形成氢键的几率,可以看出D257与3-oxo-C10-AHL之间氢键形成几率约为30%~50%,然而与3-oxo-C6-AHL之间形成氢键的几率不到20%.由此可见,长链内酯比短链内酯底物与VmoLac之间的结合能力更强.

Table 2 Hydrogen bond probability between Asp257 and polarized water molecules/3-oxo-C10-AHL/3-oxo-C6-AHL
表3比较了VmoLac/γ-nonalacton和VmoLac/ethyl-paraoxon体系中氢键形成几率的差异.其分析原理同表2,对于VmoLac/γ-nonalacton复合物,D257可以与水分子形成更强的氢键作用,即对γ-nonalacton的羰基碳具有更强的亲核进攻作用,而对于VmoLac/ethyl-paraoxon体系,paraoxon的磷原子亲核进攻作用较弱.所以VmoLac水解γ-nonalacton的能力更强.通过比较两种复合物中D257与底物之间形成氢键几率的分析,可以看出壬内酯比对氧磷底物与VmoLac之间的结合能力更强,内酯比对氧磷更适合作为VmoLac的底物.

Table 3 Hydrogen bond probability between Asp257 and polarized water molecules/γ-nonalacton/ethyl-paraoxon
3 结 论
通过对4种蛋白质复合物VmoLac/3-oxo-C10-AHL,VmoLac/3-oxo-C6-AHL,VmoLac/γ-nonalacton,VmoLac/ethyl-paraoxon进行分子动力学模拟,比较了VmoLac催化水解长链内酯、短链内酯、壬内酯以及对氧磷的能力.结果表明,在VmoLac/3-oxo-C10-AHL和VmoLac/γ-nonalacton体系中,活性口袋附近的Loop8结构域运动明显,更加柔性的构象使底物更加容易靠近Loop8疏水通道外侧.Y98与长链内酯的羰基碳之间形成的进攻距离小于短链内酯与其形成的距离,并且与壬内酯的羰基碳之间的距离小于与对氧磷的磷原子之间形成的距离,短的距离更有利于发生亲核进攻反应.D257是引发VmoLac催化反应的关键残基,其可与VmoLac/3-oxo-C10-AHL和VmoLac/γ-nonalacton体系的极化水以及底物形成更多的氢键,使底物与酶更容易结合.从理论角度解释了VmoLac催化长链内酯比短链内酯的能力强,催化内酯比对氧磷能力强的原因.所得结果为研究VmoLac与其4种非特异性底物的相互作用提供了新的思路.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210136.
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
昆明医科大学学报(2021年4期)2021-07-23 01:21:44
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
池州学院学报(2015年3期)2016-01-05 01:13:04
中国塑料(2015年10期)2015-10-14 01:13:16
天津科技大学学报(2015年2期)2015-08-09 01:40:42
医学研究杂志(2015年4期)2015-06-10 06:42:43
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
遗传(2014年3期)2014-02-28 20:59:04
