
CaMKⅡ调控脂多糖诱导的心肌细胞内钙、ROS的作用机制研究
2021-07-17 07:34:42巩奇明李德慧龚元勋姜艳
右江民族医学院学报 2021年3期
巩奇明,李德慧,龚元勋,姜艳
(1. 右江民族医学院附属医院,广西 百色 533000;2. 右江民族医学院科学实验中心,广西 百色 533000)
脓毒症是临床常见的炎症反应综合征。心肌炎是该综合征常见的并发症之一,可导致心力衰竭以及患者死亡[1]。心肌炎主要表现为细胞内线粒体活性氧(reactive oxygen species,ROS)增加,炎症因子大量分泌,细胞内钙超载和细胞凋亡等[2]。目前,对心肌炎发病机制的研究仍不够全面和深入。
钙调蛋白依赖性蛋白激酶Ⅱ(CaMK Ⅱ)是一种多功能的丝氨酸/苏氨酸激酶[3],也是钙/钙调蛋白依赖性蛋白(Ca2+/CaM)信号通路的关键调节酶[4],参与心肌细胞死亡、心肌肥大、炎症反应、缺血再灌注以及心律失常等多种病理过程[3-8]。但是目前CaMK Ⅱ在脂多糖(lipopolysaccharide,LPS)诱导的心肌细胞损伤中的分子调控机制尚不明确。
本研究采用LPS(细胞炎症反应和凋亡的主要启动因子)构建体外心肌细胞炎症模型,通过观察抑制CaMK Ⅱ是否对LPS所致心肌细胞胞浆钙含量及ROS水平具有调控作用,从中探讨CaMK Ⅱ在该作用中的具体分子机制。
1 材料与方法
1.1 材料
1.1.1 细胞株 大鼠心肌细胞(H9C2)购自上海中乔新舟生物公司。
1.1.2 主要试剂 高糖型DMEM培养基(C1199-5500BT,Gibco);胎牛血清(FSP500,ExCell Bio);qPCR SYBR Green Master Mix (11201ES08,翊圣);RT First Starand cDNA 第一链合成试剂盒(D71-68M,碧云天);LPS (L2880,Sigma);PCR引物合成(上海捷瑞);EDTA-胰蛋白酶(25200056,Gibco);KN93(HY-15465,MCE);钙离子荧光探针(Fluo-3 AM,40703ES50,翊圣);Hanks平衡盐溶液(HBSS,60161ES76,翊圣);活性氧ROS试剂盒(s0033s,碧云天);多克隆兔抗CaMK Ⅱ、单克隆鼠抗GAPDH、山羊抗兔IgG (H+L) HRP、山羊抗鼠IgG (H+L) HRP (AF6434、T0001、S0001、S0002、Affinity Biosciences)、BCA试剂盒、SDS-PAGE凝胶试剂盒、ECL化学发光溶液均购自上海雅酶公司。
1.2 细胞分组与建模 在37℃,5% CO2条件下,使用含有10%胎牛血清的DMEM培养基培养细胞。待细胞生长至80%~90%时,消化,离心,重悬,计数,接种于孔板或平皿中(接种密度6孔板为2×105个/孔;共聚焦培养皿为5×104个/平皿;30 mm直径细胞培养皿为2×106个/平皿)。继续培养24 h后将细胞分为对照组、LPS组、KN93 + LPS组。对照组不做特殊处理;LPS组:采用10.0 μg/ml的LPS作用于细胞48 h;KN93 + LPS组:先用5.0 μmol/L的KN93作用于细胞1 h,随后加入10.0 μg/ml的LPS刺激细胞48 h。
1.3 标本采集及检测
1.3.1 细胞胞浆Ca2+荧光检测 LPS作用结束后,取共聚焦培养皿中的细胞,弃除细胞培养基,HBSS溶液清洗细胞3次,使用5.0 μM的Flou-3 AM工作液,37℃孵育45 min。随后去除工作液,再次清洗细胞3次,并用HBSS溶液覆盖细胞,37℃继续孵育20 min。最后采用激光共聚焦显微镜(FV3000,日本Olympus)扫描成像。
1.3.2 细胞内ROS检测 取处理后的细胞,加入200 μl浓度为10.0 μmol/L的荧光探针DCFH-DA,37℃孵育20 min。使用DMEM培养基清洗细胞3次,随后采用激光共聚焦显微镜采集图像。
1.3.3 RT-qPCR实验 取药物处理后的细胞,清除上清液,每个平皿加入1.0 ml TRIzol试剂提取细胞内的总RNA,使用微量紫外分光光度计(TGEM Plus,北京天根)测定RNA浓度及OD260/OD280值。采用反转录试剂盒进行反逆录,获得cDNA单链;使用SYBR Green Master Mix试剂在qPCR仪(Light Cycler 9600,德国罗氏)进一步扩增样本中的cDNA。各基因引物序列见表1。扩增条件为:95℃,10 min;95℃,10 s;60℃,20 s;72℃,30 s。共计45个循环。样本均设有3个复孔。采用2-ΔΔCt方法计算目的基因的相对含量。

表1 RT-qPCR实验各基因引物序列
1.3.4 Western Blot实验 取处理后的细胞培养皿,弃除细胞上清液,并用4℃的PBS清洗细胞2次。向培养皿中加入400 μl RIPA细胞裂解液(内含1% PMSF),提取细胞总蛋白。50 μg蛋白样本用于聚丙烯酰胺凝胶(SDS-PAGE)电泳,湿转法转移蛋白至孔径为0.45 μm的PVDF膜上,室温封闭10 min,使用1∶1000稀释后的一抗溶液4℃孵育过夜。在1∶2000的HRP标记的山羊抗兔(或抗鼠)IgG抗体溶液中室温孵育50 min。使用ECL发光溶液,进行曝光成像。采用ImageJ软件对条带进行灰度值分析,计算心肌细胞中CaMK Ⅱ蛋白表达水平。

2 结果
2.1 抑制CaMK Ⅱ可降低LPS所致细胞胞浆Ca2+增加 荧光探针检测结果显示:经LPS处理后细胞胞浆Ca2+水平升高,且高于对照组。而在KN93+LPS组细胞中LPS对Ca2+的增加作用被抑制。见图1。

图1 各组心肌细胞胞浆Ca2+含量检测(800 ×,400 ×)
2.2 抑制CaMK Ⅱ可降低LPS诱导的细胞内ROS增加 分析ROS荧光图:与对照组比较,LPS组细胞内ROS含量增加,表现为荧光强度强于对照组。而KN93预处理的KN93 + LPS组细胞中,ROS水平低于LPS组。见图2。
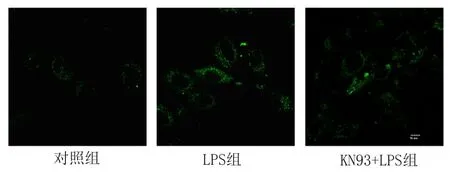
图2 各组心肌细胞内ROS含量检测(800×)
2.3 细胞内CaMK Ⅱ、受磷蛋白(PLB)、心肌细胞肌浆网Ca2+-ATP酶(SERCA)、无翅型小鼠乳腺肿瘤病毒整合位点家族成员4(Wnt4)、β-连环蛋白(β-catenin)、原癌基因(c-Myc)、G1/S-特异性周期蛋白D1(Cyclin D1)、诱导型一氧化氮(iNOS)、白介素10(IL-10)、mRNA表达水平。
2.3.1 KN93降低LPS对CaMK Ⅱ的促进作用 RT-qPCR和Western Blot实验结果显示:LPS能显著增加心肌细胞中CaMK Ⅱ mRNA和蛋白的表达,与对照组比较差异有统计学意义(P<0.01)。而在KN93+LPS组,KN93预处理显著降低CaMK Ⅱ的mRNA和蛋白水平,与LPS组比较差异有统计学意义(P<0.01)。见图3。

注:与对照组比较,** P<0.01;与LPS组比较,##P<0.01。图3 各组心肌细胞内CaMK ⅡmRNA(A)和蛋白(B)水平检测
2.3.2 抑制CaMK Ⅱ可缓解LPS对PLB和SERCA mRNA的抑制作用 LPS组细胞受LPS影响,细胞内钙调控蛋白PLB和SERCA mRNA表达水平降低,与对照组相比差异有统计学意义(P<0.01)。而在KN93+LPS组,PLB和SERCA mRNA表达升高,与LPS组比较差异有统计学意义(P<0.01)。见图4。
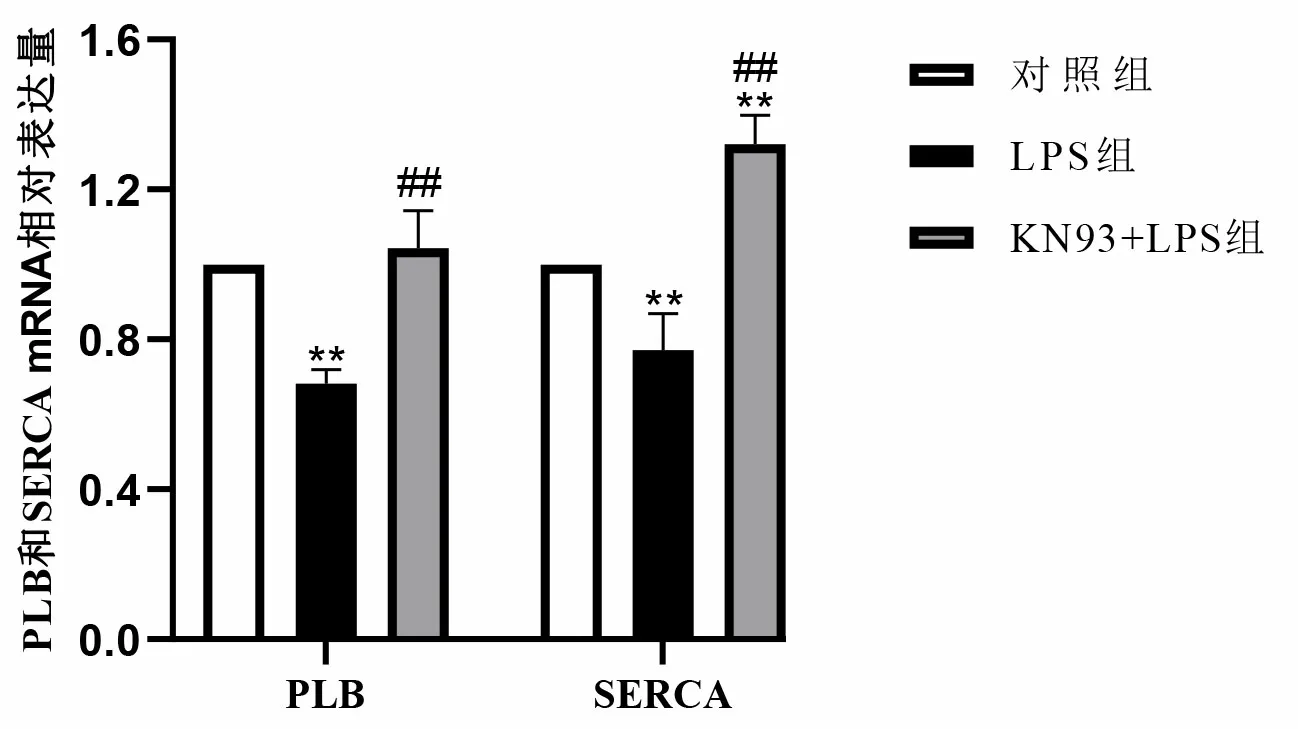
注:与对照组比较,** P<0.01;与LPS组比较,##P<0.01。图4 各组心肌细胞内PLB和SERCA mRNA水平检测
2.3.3 抑制CaMK Ⅱ对LPS诱导的Wnt4和β-catenin mRNA水平的影响 LPS组细胞受LPS影响,Wnt4和β-catenin mRNA表达水平升高,且高于对照组(P<0.05或0.01)。对比于LPS组,KN93 + LPS组细胞内Wnt4 mRNA表达水平的降低(P<0.05),而β-catenin mRNA含量水平无明显变化(P>0.05)。见图5。
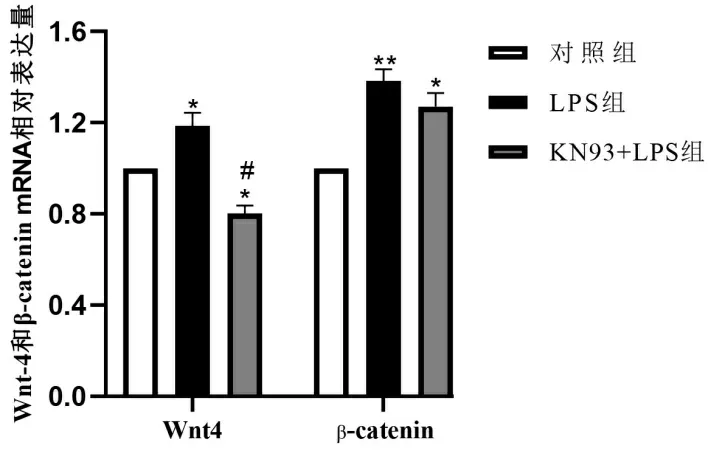
注:与对照组比较,** P<0.01,* P<0.05;与LPS组比较,##P<0.01,#P<0.05。图5 各组心肌细胞内Wnt4和β-catenin mRNA水平检测
2.3.4 抑制CaMK Ⅱ对LPS诱导的c-Myc和Cyclin D1 mRNA水平的影响 LPS组细胞内c-Myc和Cyclin D1的mRNA水平显著增加,对比于对照组差异有统计学意义(P<0.01)。而在KN93 + LPS组,KN93可降低LPS对Cyclin D1 mRNA的促进作用(对比LPS组,P<0.05)。见图6。
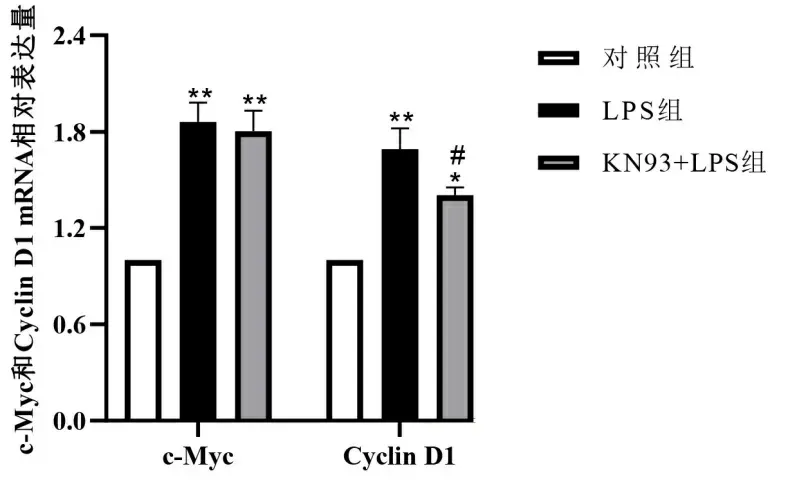
注:与对照组比较,** P<0.01,* P<0.05;与LPS组比较,#P<0.05。图6 各组心肌细胞内c-Myc和Cyclin D1 mRNA水平检测
2.3.5 抑制CaMK Ⅱ可降低LPS诱导的iNOS和IL-10 mRNA水平 对比于对照组,LPS组细胞内iNOS和IL-10 mRNA水平显著增加(P<0.01)。而KN93+LPS组内iNOS和IL-10 mRNA水平低于LPS组(P<0.05或<0.01)。见图7。
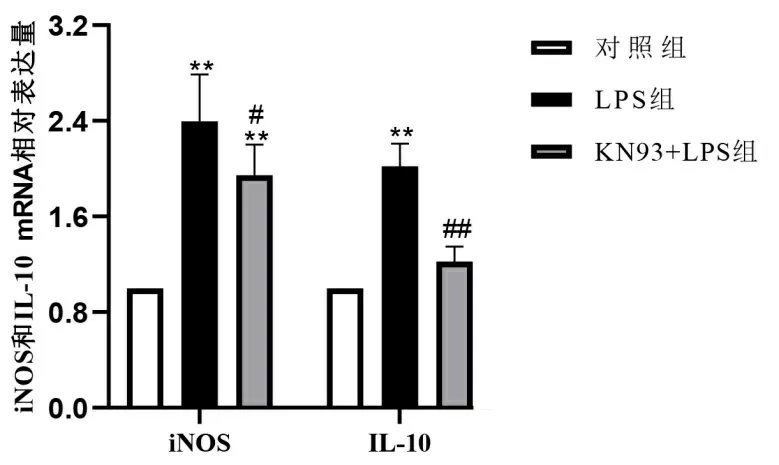
注:与对照组比较,** P<0.01;与LPS组比较,##P<0.01,#P<0.05。图7 各组心肌细胞内iNOS和IL-10 mRNA水平检测
3 讨论
心肌细胞胞浆中的Ca2+含量受内质网和线粒体共同调控[6],同时也受多种钙离子通道以及钙离子调控相关信号通路的共同影响。心肌细胞被LPS损伤后,细胞内受磷蛋白PLB功能下降,肌质网摄取钙减少,细胞膜上的钠钙交换减少,细胞胞浆Ca2+不能及时降低至正常水平,造成细胞胞浆钙超载,导致钙稳态失衡[9],损伤心肌细胞兴奋收缩功能[1-2]。研究表明[6],KN93通过抑制CaMK Ⅱ的活性并降低其磷酸化水平,从而对CaMK Ⅱ发挥特异性抑制作用。研究证实,KN93抑制CaMK Ⅱ,降低缺血再灌注损伤钙波的产生,从而降低再灌性心律失常的发生。KN93抑制CaMK Ⅱ活性,可有效降低LPS所致细胞内钙超载,与上述结果一致。
此外,LPS诱导细胞内ROS过度增加,破坏细胞膜结构[10],诱发内质网应激和细胞凋亡[11]。本研究表明,CaMK Ⅱ表达降低可减少细胞ROS含量,防止ROS对心肌细胞进一步损伤。
Wnt信号通路有Wnt经典通路和Wnt非经典通路两种。Wnt经典通路,又称Wnt/β-catenin信号通路,具有调控细胞生长、增殖以及凋亡的作用。Wnt/β-catenin信号通路可作为心血管疾病发生发展的生物标志物,参与心肌损伤及修复调控。在LPS损伤心肌细胞内可检测到Wnt/β-catenin表达增加[12],β-catenin从细胞膜上不断进入细胞浆,并在胞浆内大量聚集。当β-catenin浓度过高时,β-catenin进入细胞核,激活下游细胞周期素D1(cyclin D1)和C-myc等细胞肥大相关基因,最终导致心肌细胞肥大性病变[13]。Wnt非经典通路又分为Wnt/Ca2+通路和Wnt/JNK通路。Wnt/Ca2+通路下游信号分子有磷脂酶C、钙离子、CaMK Ⅱ等分子。胞浆Ca2+增加可激活CaMK Ⅱ进而促进细胞核内肥大相关蛋白的活性,进一步加速心肌肥大病程[13]。Wnt/JNK通路下游有C-Jun氨基末端激酶(JNK)和细胞核内转录因子C-Jun等分子[13],该通路的激活与心肌肥大、纤维化等病理过程相关。本研究观察到LPS损伤的心肌细胞中,Wnt/β-catenin、c-Myc/CyclinD1的活性均增加;而抑制CaMK Ⅱ表达,可显著抑制Wnt4、CyclinD1 mRNA的表达水平。
作为非Ca2+依赖型NOS亚型,iNOS受LPS刺激后大量增加,并能够持续性大量释放NO[14]。此外,暴露于LPS的心肌细胞可分泌大量炎性因子(TNF-a、IL-1β以及IL-6等)[15-16]。本研究发现抑制CaMK Ⅱ的表达可抑制iNOS、IL-10转录,降低炎性反应的进一步发展。综上所述,本研究发现CaMK Ⅱ通过调控心肌细胞内钙调蛋白PLB、SERAR,Wnt通路的Wnt4、CyclinD1,以及炎症通路的iNOS、IL-10的基因表达水平,减轻LPS所致的心肌细胞内Ca2+和ROS水平增加。这表明CaMK Ⅱ可能是脓毒症心肌炎的一个潜在治疗靶点。但本文的实验结果尚不够完善,还需要体内实验进一步验证。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:16:04
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
阅读(科学探秘)(2021年9期)2021-05-30 10:48:04
考试与评价·高二版(2020年4期)2020-09-10 07:22:44
橡塑技术与装备(2018年14期)2018-07-20 03:22:00
销售与市场(管理版)(2017年3期)2017-03-28 05:59:25
海南医学(2016年8期)2016-06-08 05:43:00
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:56
中国男科学杂志(2016年9期)2016-03-20 15:00:13
中国病理生理杂志(2015年8期)2015-12-21 12:38:08
