
低氧诱导视网膜色素上皮细胞损伤的生物学机制转录组测序分析
2021-07-16 07:23:50卢聪史平玲杨琪翔宋昊李苗张贝贝宋宗明
中华实验眼科杂志 2021年6期
卢聪 史平玲 杨琪翔 宋昊 李苗 张贝贝 宋宗明
河南大学人民医院 河南省人民医院眼科 河南省立眼科医院 河南省眼科研究所,郑州 450003
持续的视网膜缺血、缺氧会造成组织细胞损伤,引起细胞凋亡或坏死,进而导致糖尿病视网膜病变、视网膜中央静脉阻塞和早产儿视网膜病变等疾病的发生和发展,造成严重的视力损害[1]。研究表明,缺氧能够引起视网膜色素上皮(retinal pigment epithelium,RPE)细胞以及视网膜血管功能的紊乱,在缺氧环境中RPE细胞会持续表达血管内皮生长因子(vascular endothelial growth factor,VEGF)[2-4],促使视网膜及脉络膜微血管内皮细胞不断分裂增生,形成异常新生血管,继而引起视力的不可逆损害。因此,减缓甚至阻止视网膜缺血缺氧进程变得尤为重要。低氧诱导因子(hypoxia-inducible factors-1α,HIF-1α)是一种重要的核转录调节因子,在缺氧条件下被激活,并诱导包括VEGF在内的多种基因产物的表达[5]。RPE细胞位于神经视网膜外的色素细胞层,与上层视网膜感光细胞和下层脉络膜紧密相连并滋养视网膜感光细胞,在缺血或低氧状态下极易受到影响并造成损伤[2,6-7]。深入探讨低氧诱导RPE细胞损伤的分子机制有助于相关疾病的早期防治,改善患者预后。转录组测序技术又称RNA测序(RNA sequencing,RNA-seq),具有高通量、全方位、快速获取转录本的特点[8-9]。用于定量基因在细胞、组织、器官乃至整个机体中的表达异质性,在破译基因组结构和功能、识别细胞生物系统下的遗传网络、建立应对疾病、病原体的分子生物标志物等方面发挥着重要作用[10-12]。生物信息学技术是采用计算机技术结合信息论方法对蛋白质、核酸信息进行采集、存储、传递以及分析的科学,通过数据库建设、序列分析、结构分析与功能预测、大规模功能表达谱分析、代谢网络建模分析等,整合数据量巨大的核酸、蛋白质信息,使之成为具有明确生物学意义的生物信息[13-14]。本实验拟通过RNA-seq及生物信息学技术分析低氧与常氧处理下的ARPE-19细胞基因表达谱变化,探讨低氧诱导的RPE细胞损伤发生和发展可能的分子机制。
1 材料与方法
1.1 材料
1.1.1细胞来源 ARPE-19细胞购自美国ATCC公司。
1.1.2主要试剂及仪器 胎牛血清(fetal bovine serum,FBS)(11011-8611,浙江天杭生物科技股份有限公司);DMEM-F12培养基、胰蛋白酶-EDTA溶液(北京索莱宝科技有限公司);Trizol®试剂(美国Thermo Fisher公司);逆转录试剂盒PrimeScriptTMRT reagent kit with gDNA Eraser、实时荧光定量PCR试剂盒TB Green®premix Ex TaqTMⅡ试剂(日本Takara Biotechnology公司);实时荧光定量PCR引物(上海生工生物工程股份有限公司);细胞活力(活死细胞染色)检测试剂盒(北京百奥莱博科技有限公司)。细胞培养箱(美国Thermo Fisher公司);Stepone Plus实时荧光定量PCR仪(美国Applied Biosystems公司);荧光显微镜(德国Carl Zeiss公司)。
1.2 方法
1.2.1细胞培养 将保存于液氮中的ARPE-19细胞置于37 ℃恒温水浴锅中解冻复苏,待细胞融化后快速转移至含体积分数10% FBS的DMEM-F12培养基15 ml的离心管中混匀,室温下156×g离心5 min,弃上清,用1 ml培养基悬浮细胞沉淀,吹打均匀后将细胞置于25 cm2细胞培养瓶中,每瓶中加入3~4 ml培养基进行培养。待ARPE-19细胞密度达到80%~90%进行传代培养,弃去培养基,使用磷酸盐缓冲液(phosphate buffer saline,PBS)清洗2次。每瓶加入1 ml质量分数0.25%的胰蛋白酶-EDTA消化液,放入细胞培养箱消化2 min取出。每瓶加入1 ml DMEM-F12培养基终止胰蛋白酶消化,转移至15 ml离心管,156×g离心5 min,弃上清液。加入DMEM-F12培养基吹匀,进行细胞计数,以2×107/皿细胞的密度将ARPE-19细胞接种至直径10 cm培养皿中,并置于37 ℃、体积分数5% CO2培养箱中孵育,待细胞贴壁生长后将细胞分为常氧对照组和低氧处理组,分别置于体积分数21%和1% O2的三气培养箱中继续培养8、24、48和72 h,收集细胞,以进行后续实验。
1.2.2RNA-seq及生物信息学分析 低氧处理组与常氧对照组各设6个细胞生物学重复样本,于处理后8 h和24 h各组各收集3个细胞样本,使用2 ml/皿Trizol®试剂进行处理,每个样品取1 ml送至武汉华大医学检验所提取总RNA,通过Fragment Analyzer进行浓度和质量检测,质量合格的RNA使用BGISEQ-500平台进行RNA-seq,测序长度为SE50,绝大多数转录本被完整覆盖,同时reads均匀分布在转录本的各个区域。原始数据(raw reads)以FASTQ格式记录,经Trimmomatic软件过滤掉包含接头的reads(接头污染)、未知碱基N含量大于5%的reads(N表示无法确定的碱基信息)、低质量reads(质量值Q<10的碱基数占整个reads总碱基的比例大于20%以上的reads),得到高质量clean reads,以保证结果的可靠性。使用HISAT将clean reads比对到参考基因组序列,使用Bowtie2将clean reads比对到参考基因序列,使用标准化算法FPKM(fragments per kilo-base per million mapped fragments),即每百万个片段中比对上某转录本每千碱基长度的片段数目作为基因表达水平的衡量指标。本研究将|log2FC|≥1、P≤0.05的基因作为筛选差异表达基因(differentially expressed genes,DEGs)的标准,对DEGs进行生物信息学分析。用火山图描绘低氧刺激ARPE-19细胞8 h和24 h后DEGs的分布情况。使用R语言中的pheatmap包对DEGs进行基因间与样本间的聚类热图分析。每列代表1个样品,每行代表1个转录本,颜色越红表达量越高,颜色越绿表达量越低。采用Dr.TOM系统对DEGs进行基因本体论(Gene Ontology,GO)功能注释分析及京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)信号通路富集分析,筛选出参与调控低氧反应的信号通路,采用String数据库和Cytoscape软件构建ARPE-19细胞处理24 h后DEGs的蛋白-蛋白互作(protein-protein interaction,PPI)网络图,寻找参与低氧反应的功能性靶基因。连线代表基因间的调控关系。颜色越亮、图标越大代表该基因在网络中与其他基因关系越密切,功能越重要。

表1 2个组DEGs引物序列Table 1 Primer sequence of DEGs in the two groups基因 NM.序列号引物序列(5’-3’)log2FCDEPP1NM_145980.2正向:GTGAGGTCTATATCTCGACTGGC反向:ACTGAAACGTGCGGTGATGT1.574349NPPBNM_008726.6正向:TGGAAACGTCCGGGTTACAG反向:CTGATCCGGTCCATCTTCCT1.419870PDZK1NM_002614.4正向:CATGATCCTGACCGTCGGAAA反向:TGCTCACTGGACCTGAAACTG1.452112HILPDANM_001193365.1正向:AAGCATGTGTTGAACCTCTACC反向:TGTGTTGGCTAGTTGGCTTCT1.397986NDRG1NM_001135242.2正向:CTCCTGCAAGAGTTTGATGTCC反向:TCATGCCGATGTCATGGTAGG1.260060RORCNM_005060.4正向:CTGGGCATGTCCCGAGATG反向:GAGGGGTCTTGACCACTGG1.836842TCEA3NM_003196.3正向:CCCCAAAACACCTAGCAGC反向:CTTCATGTCCGTGCTCTTGAG1.230815TFRCNM_011638.4正向:GGCTACTTGGGCTATTGTAAAGG反向:CAGTTTCTCCGACAACTTTCTCT-2.138634NQO1NM_000903.3正向:GAAGAGCACTGATCGTACTGGC反向:GGATACTGAAAGTTCGCAGGG-1.101194 注:DEGs:差异表达基因;FC:倍数变化 Note:DEGs:differentially expressed genes;FC:fold change
1.2.3实时荧光定量PCR法检测与低氧相关基因的mRNA表达水平 将1.2.1中收集的ARPE-19细胞使用Trizol法提取细胞的总RNA,经Thermo SPECTRONIC 200分光光度计检测其浓度和吸光度(A)值。采用逆转录试剂盒将RNA逆转录成cDNA,实时荧光定量PCR定量VEGF、HIF-1α mRNA表达量,预实验以对低氧刺激较为敏感的VEGF和HIF-1α在mRNA水平上发生的变化为条件筛选合适时间点进行RNA-seq。根据预实验中不同时间点mRNA表达量的差异结果并结合相关文献报道[4,15-16],选择mRNA表达差异较大的8 h、24 h这2个时间点,收集样本后进行RNA-seq。根据测序结果和文献报道,筛选DEGs中可能与低氧相关的9个基因DEPP1、NPPB、PDZK1、HILPDA、TCEA3、NDRG1、RORC、TFRC和NQO1,验证高通量测序结果的准确性。设计9个基因的正向引物和反向引物(表1),以β-actin为内参基因。使用TB Green®Premix Ex TaqTMⅡ试剂盒进行实时荧光定量PCR反应;用于定量的反应体系包括TB Green®Premix Ex Taq Ⅱ 10.0 μl,ROX reference Dye 0.4 μl,正向引物和反向引物各2.0 μl,cDNA模板5.6 μl,共20.0 μl。反应条件:95 ℃预变性30 s;95 ℃变性5 s,60 ℃退火,延伸30 s,40个循环;同一样品设置3个重复孔。采用2-△△CT法计算各个基因的mRNA相对表达水平。
1.2.4细胞活性荧光染色检测低氧环境下细胞生存状态 取处于对数生长期的ARPE-19细胞,接种至24孔细胞培养板中,每孔2×104个细胞,置于37 ℃、5% CO2、1% O2培养箱孵育8、24、48、72 h后,弃去培养基,PBS洗涤2次,使用细胞活力检测试剂盒按照说明书向孔内加入检测试剂进行染色,使用ImageJ软件分析及荧光显微镜成像观察低氧处理后不同时间点细胞的生存状态。
1.3 统计学方法
采用GraphPad Prism 7软件进行统计分析。本研究中计量资料数据经Shapiro-Wilk检验证实呈正态分布,以mean±SD表示。常氧对照组与低氧处理组不同时间点VEGF、HIF-1α和DEGs mRNA相对表达量的比较采用独立样本t检验。P<0.05为差异有统计学意义。
2 结果
2.1 2个组不同时间点ARPE-19细胞中VEGF、HIF-1α mRNA相对表达量比较
低氧处理组8、24、48、72 h VEGF和低氧处理组8、24、48 h HIF-1α mRNA相对表达量均高于常氧对照组,差异均有统计学意义(均P<0.05);2个组VEGF和HIF-1α mRNA相对表达量在24 h时差异最大,其次是8 h(表2)。本实验选择8 h、24 h作为低氧处理时间,处理后收集样本进行RNA-seq。
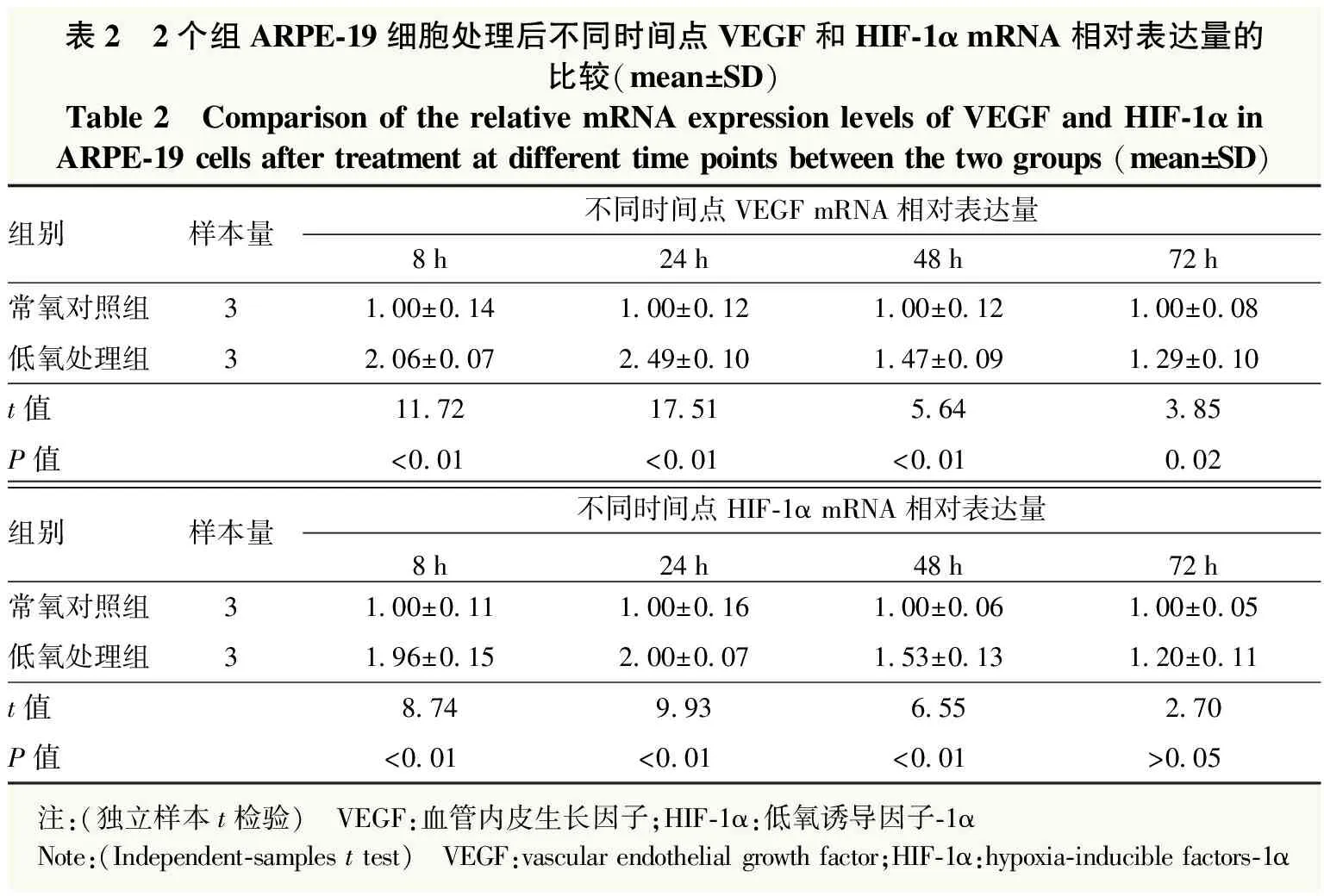
表2 2个组ARPE-19细胞处理后不同时间点VEGF和HIF-1α mRNA相对表达量的比较(mean±SD)Table 2 Comparison of the relative mRNA expression levels of VEGF and HIF-1α in ARPE-19 cells after treatment at different time points between the two groups (mean±SD)组别样本量不同时间点VEGF mRNA相对表达量8h24h48h72h常氧对照组31.00±0.141.00±0.121.00±0.121.00±0.08低氧处理组32.06±0.072.49±0.101.47±0.091.29±0.10t值11.7217.515.643.85P值<0.01<0.01<0.010.02组别样本量不同时间点HIF-1α mRNA相对表达量8h24h48h72h常氧对照组31.00±0.111.00±0.161.00±0.061.00±0.05低氧处理组31.96±0.152.00±0.071.53±0.131.20±0.11t值8.749.936.552.70P值<0.01<0.01<0.01>0.05 注:(独立样本t检验) VEGF:血管内皮生长因子;HIF-1α:低氧诱导因子-1α Note:(Independent-samples t test) VEGF:vascular endothelial growth factor;HIF-1α:hypoxia-inducible factors-1α
2.2 2个组ARPE-19细胞处理8 h、24 h后样品测序数据
常氧、低氧处理ARPE-19细胞8 h、24 h后测序共得到12组转录组数据,原始数据过滤掉测序接头序列、未知碱基序列、低质量序列后得到高质量测序数据clean reads(表3)。

表3 2个组ARPE-19细胞处理8h、24h后样品测序数据过滤后的reads质量统计Table 3 Quality statistics of filtered reads from the sequencing data after 8-hour and 24-hour treatment of ARPE-19 cells in the two groups样本名称过滤后readsreads数目(M)Q20(%)Q30(%)8h 常氧组122.8498.7895.188h 常氧组222.8498.7395.018h 常氧组322.8498.7895.328h 低氧组122.8898.8695.498h 低氧组222.8298.7695.158h 低氧组322.8798.7595.1424h常氧组125.1298.0093.9524h常氧组225.9597.6993.1724h常氧组325.9597.9093.7624h低氧组125.9597.8493.6124h低氧组223.9397.8093.5524h低氧组325.9497.7993.44 注:Q20为过滤后质量不低于20的碱基比例,Q30为过滤后质量不低于30的碱基比例 Note:Q20 was the ratio of bases with a quality of no less than 20 after fil-tration,and Q30 was the ratio of bases with a quality of no less than 30 after filtration
2.3 2个组ARPE-19细胞处理8 h、24 h后DEGs分析
低氧处理8 h和24 h后显著DEGs中有9个相同基因(图1)。低氧刺激ARPE-19细胞8 h和24 h后DEGs的分布情况见图2。低氧处理8 h,低氧处理组与常氧对照组间相比共筛选出62个DEGs,其中显著上调基因45个,显著下调基因17个。低氧处理24 h,2个组间共筛选出255个显著DEGs,其中显著上调基因228个,显著下调基因27个。

图1 2个组ARPE-19细胞处理8 h、24 h后DEGs Venn图 紫色表示处理8 h后显著DEGs的基因个数;黄色表示处理24 h后显著DEGs的基因个数;灰色表示处理8 h和24 h显著DEGs中相同的基因个数

图2 2个组ARPE-19细胞处理8 h、24 h后DEGs火山图 A:低氧处理8 h DEGs火山图 B:低氧处理24 h DEGs火山图 FC:倍数变化;DEGs:差异表达基因
2.4 2个组ARPE-19细胞处理8 h、24 h后DEGs的聚类热图分析
每组3个生物学样本重复性较好,同组样本间具有较高的相关性,2个组各时间点在整体基因表达方面存在明显差异(图3)。
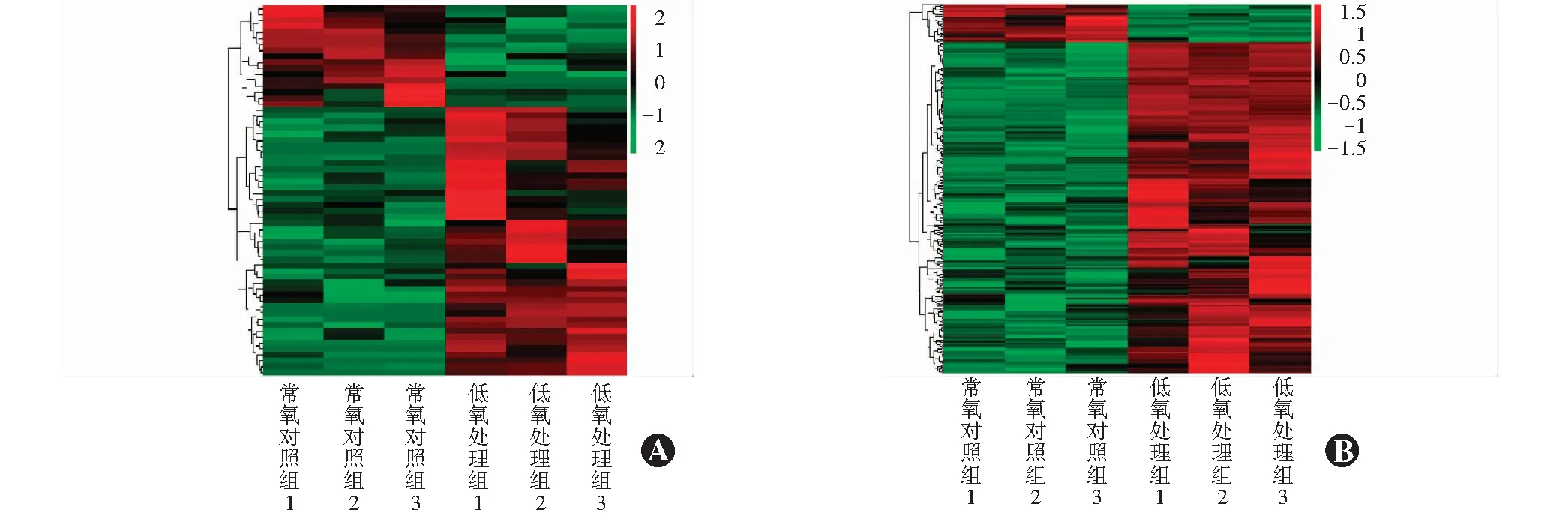
图3 2个组ARPE-19细胞低氧处理8 h、24 h后DEGs聚类热图分析 每列代表1个样品,每行代表1个转录本,颜色越红则表达量越高,颜色越绿则表达量越低,2个组在整体基因表达方面存在明显差异 A:低氧处理8 h DEGs聚类热图 B:低氧处理24 h DEGs聚类热图
2.5 2个组ARPE-19细胞处理8 h、24 h后DEGs的GO功能注释分析
低氧处理8 h后2个组DEGs没有显著富集GO功能进程,处理24 h后2个组DEGs经过GO功能富集分析在25个生物学进程条目、17个细胞组分条目、11个分子功能条目(P≤0.05)。对所有DEGs进行GO功能注释,结果显示在生物学过程中前20个富集的条目功能主要为蛋白质降解、核苷酸生物合成、超氧化物自由基清除、肌动蛋白纤维及物质转运等进程;在分子功能中,整合与催化活性条目富集程度最高;在细胞组分本体中,细胞条目富集程度最高(图4)。
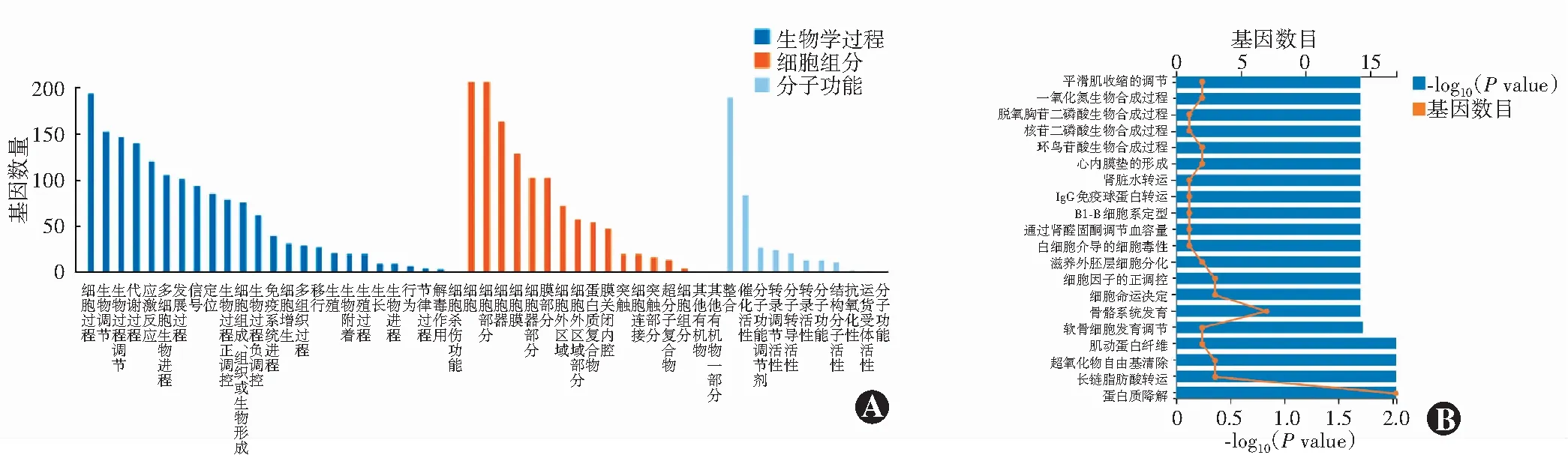
图4 2个组ARPE-19细胞处理24 h后DEGs的GO功能富集分析图 柱状图代表显著富集的GO条目(P≤0.05),折线代表该条目包含的基因个数 A:GO功能分析 B:GO生物学进程分析
2.6 2个组ARPE-19细胞处理8 h、24 h后DEGs的KEGG通路富集分析
KEGG通路富集分析结果显示,低氧处理8 h后2个组间DEGs没有KEGG显著富集通路,处理24 h后2个组间DEGs筛选出17条KEGG显著富集通路(P≤0.05),主要富集在PI3K-Akt、cGMP-PKG信号通路以及其他与代谢、细胞周期、细胞生长和细胞凋亡、铁死亡密切相关的信号通路(图5)。

图5 2个组ARPE-19细胞处理24 h后DEGs的KEGG通路富集分析 柱状图为显著富集的KEGG通路条目(P≤0.05);折线图为富集KEGG通路靶基因个数 图6 低氧处理ARPE-19细胞24 h后DEGs的PPI网络图 网络图中的连线代表基因间的调控关系,颜色越亮、图标越大代表该基因在网络中与其他基因关系越密切,功能越重要
2.7 2个组ARPE-19细胞处理24 h后DEGs的PPI网络分析
低氧处理ARPE-19细胞24 h后筛选到的DEGs通过String平台得到关键基因HPCA、MT3和NOS3,所对应的靶蛋白海马素钙结合蛋白(hippocalcin,HPCA)、金属硫蛋白3(metallothionein,MT3)、一氧化氮合酶3(nutric oxide synthase,NOS3)可能是网络中最重要的靶点蛋白(图6)。
2.8 低氧处理ARPE-19细胞24 h后与低氧相关的DEGs的验证
低氧处理ARPE-19细胞24 h后,DEPP1、NPPB、PDZK1、HILPDA、TCEA3、NDRG1和RORC的mRNA表达水平显著上调,TFRC、NQO1的mRNA表达水平显著下调(表4),与测序结果变化一致。

表4 2个组ARPE-19细胞处理24h后DEGs mRNA相对表达量的比较(mean±SD)Table 4 Comparison of relative mRNA expression levels of DEGs in ARPE-19 cells after 24-hour treatment between the two groups (mean±SD)组别样本量各基因mRNA相对表达量DEPP1NPPBPDZK1HILPDANDRG1TCEA3RORCTFRCNQO1常氧对照组31.00±0.071.00±0.211.00±0.301.00±0.061.00±0.191.00±0.041.00±0.171.00±0.131.00±0.10低氧处理组37.95±0.454.05±0.322.97±0.272.36±0.331.79±0.031.53±0.204.13±0.590.29±0.010.27±0.03t值26.1913.51 8.39 6.95 7.15 4.42 8.84 9.13 7.62P值<0.01<0.01<0.01<0.01<0.01<0.05<0.01<0.01<0.01 注:(独立样本t检验) DEGs:差异表达基因 Note:(Independent-samples t test) DEGs:differentially expressed genes
2.9 低氧处理ARPE-19细胞不同时间点活性荧光染色细胞状态变化
低氧处理ARPE-19细胞8 h、24 h后,细胞形态均正常,生长状态较好,未出现死亡细胞,低氧处理48 h后ARPE-19细胞出现死亡,低氧处理72 h后死亡细胞数量增加(图7)。

图7 低氧诱导后不同时间点ARPE-19细胞活性荧光染色(×200,标尺=100 μm) 8 h、24 h时,细胞无死亡;48 h后细胞出现死亡;72 h后死亡细胞数量显著增加 蓝色荧光为Hochest33342标记细胞核;红色荧光为PI标记已破膜的死细胞;绿色荧光为Calcein-AM标记具有细胞活性的细胞
3 讨论
缺血、缺氧影响视网膜疾病的发生和发展,是许多眼病共同的病理特征[17-18]。在细胞水平上,缺血、缺氧诱导的视网膜损伤包括线粒体酶活性降低、钙稳态损伤、能量衰竭和缺氧引起的氧化应激[1]。因此,针对低氧诱导细胞损伤的研究对于恢复正常的视网膜环境、干预和防止视网膜不可逆损伤至关重要。本实验采用RNA-seq和生物信息学分析方法研究低氧诱导的ARPE-19细胞损伤的分子机制,为研究缺血、缺氧性视网膜疾病的病理过程提供实验依据。
近年来随着RNA-seq的迅速发展,其被广泛地应用于基因表达及调控的研究中[19-20]。本研究发现,cGMP-PKG信号通路在低氧条件下参与细胞活化及抗氧化反应,对细胞起到保护作用。研究表明,NO-cGMP-PKG通路的激活被认为是抑制细胞凋亡的重要途径[21];PI3K-Akt是HIF-1α的主要上游调控因子之一,在低氧损伤进程中发挥了关键作用[22]。低氧环境下,PI3K-Akt信号通路被激活,通过活化生存相关的蛋白和失活凋亡相关的蛋白调节HIF-1α的表达来发挥功能[23-24],HIF-1α又可进一步调控下游VEGF变化,促进缺血适应[4,25]。研究表明,通过PI3K/Akt通路可减弱氧化应激诱导的细胞损伤[26]。VEGF是一种重要的生长因子,与细胞和新生血管生长密切相关[3]。HIF-1α作为适应缺氧反应的主要转录调节因子,缺氧条件下其表达量升高,能够激活多个基因的转录、增加蛋白产物氧输送、促进代谢进程以适应低氧环境。HIF-1α和VEGF均为PI3K-Akt低氧相关信号通路的下游因子[23],低氧可激活PI3K-Akt通路,VEGF和HIF-1α mRNA高表达是低氧诱导细胞损伤模型建立的关键指标。
本研究进行PPI网络分析发现了处于核心位置的关键基因HPCA、MT3和NOS3,其中HPCA基因编码的蛋白质是视网膜和大脑中发现的神经元特异性钙结合蛋白家族成员,在光感受器细胞中以钙敏感方式调节光信号转导。而低氧环境下,细胞内的钙稳态极易被破坏,因此HPCA基因的表达易受缺氧的影响[27];MT3基因在缺氧环境中易被诱导而表达升高,参与细胞抗氧化应激和凋亡,该基因表达的蛋白可保护细胞免受低氧损伤[28];NOS3基因主要参与精氨酸和脯氨酸的代谢及氧化应激,可以促进NO的产生,通过cGMP介导的信号转导通路参与血管平滑肌松弛过程,在缺血、缺氧时,通过调节活性氮与活性氧来减少细胞因缺血受到的损伤[29]。这些基因的功能主要与应对氧化应激相关。
本研究中低氧处理8 h与24 h测序重合的9个基因中,除TFRC、RORC基因可能参与低氧反应,其余基因有关低氧方面的研究较少,与低氧诱导细胞损伤、代谢进程等无明显直接关系,因此本研究中选择可能与低氧相关的基因进行实时荧光定量PCR验证,实验结果与测序结果一致。其中黄体酮诱导的蜕膜蛋白由DEPP1基因编码,参与低氧反应与自噬等细胞途径的激活,其表达受氧化应激调控[30];RORC基因可有效抑制HIF-1α的活性,并调控其转录表达,抑制肿瘤细胞的增生[31];NDRG1在缺氧条件下表达明显上调,在调节细胞分化、增生、凋亡、血管生成、肿瘤进展方面发挥重要作用[32];HILPDA是一种与缺氧诱导脂滴形成相关的基因,参与肝细胞、脂肪细胞、巨噬细胞以及肿瘤细胞等脂质代谢进程[33];NPPB、PDZK1和TCEA3也多参与代谢、细胞的生长与分化等进程。TFRC是参与铁代谢的关键因子[34],参与铁死亡过程,低氧环境下TFRC mRNA的表达受HIF-1α的调节。因此进一步研究铁死亡信号通路在低氧诱导细胞损伤中的作用,将成为我们后续研究的重点之一。
本实验不仅在分子水平上进行验证,还对低氧处理后ARPE-19细胞状态的变化进行表型研究。本实验推测,24~48 h的细胞可能处于对数生长期,此时细胞增生较多,密度增加;随着缺氧时间的延长,细胞活力逐渐下降,至48 h出现细胞死亡,此时细胞增生能力也逐渐降低,细胞密度缓慢增加。本实验方案起始状态细胞密度为2×104个/孔,相对较少。随着观察时间的延长,细胞会有不同程度的增生。此时会出现细胞活性减弱,密度增加的现象。
本研究方法学的局限性:(1)缺血、缺氧性视网膜疾病发病机制复杂,体外细胞实验的培养环境及培养条件不能完全模拟体内缺氧情况,还需通过动物实验进一步验证;(2)采用生物信息学方法分析低氧相关基因及信号通路的改变,但未对这些基因及信号通路做更深入的研究,仍需要进一步实验探索。
综上所述,本研究推测cGMP-PKG、PI3K-Akt、细胞凋亡、铁死亡信号通路参与了ARPE-19细胞的低氧反应,DEPP1、NPPB、PDZK1、HILDA、TCEA3、NDRG1、RORC、TFRC和NQO1基因可能在ARPE-19细胞应对低氧损伤中起到关键作用。HPCA、MT3和NOS3基因可作为研究缺血、缺氧相关眼底病的潜在功能性靶基因。这些通路及基因与缺氧诱导的细胞损伤有一定相关性,在视网膜缺血、缺氧疾病的病理过程中发挥重要作用,有望成为治疗缺血、缺氧性视网膜疾病的新靶点。
利益冲突所有作者均声明不存在任何利益冲突
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11 09:53:56
新民周刊(2022年27期)2022-08-01 07:04:49
中医眼耳鼻喉杂志(2021年1期)2021-07-22 07:38:28
天津医科大学学报(2021年3期)2021-07-21 09:03:42
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:34
传染病信息(2021年6期)2021-02-12 01:52:58
湖南中医药大学学报(2016年1期)2016-12-01 04:08:18
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:34
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
