
大分舌蜂不同龄期幼虫及滞育预蛹肠道细菌群落多样性及其差异
2021-07-15 12:33:50寇若玫窦飞越周泽扬黄敦元
昆虫学报 2021年6期
寇若玫, 李 月, 窦飞越, 周泽扬, 黄敦元
(重庆师范大学生命科学学院, 媒介昆虫重庆市重点实验室, 重庆 401331)
社会性群居蜜蜂中,工蜂肠道中核心微生物主要涉及到变形菌门(Proteobacteria)中的Snodgrassellaalvi和Gilliamellaapicola(Kwong and Moran, 2012)、厚壁菌门(Firmicutes)中的乳杆菌属LactobacillusFirm-4和Firm-5(Babendreieretal., 2007; Martinsonetal., 2011)、放线菌门(Actinobacteria)中的Bifidobacteriumasteroides(Bottacinietal., 2012)5个物种分支。而在独栖蜜蜂的相关研究表明肠道中同样存在稳定的核心菌群,包括:醋酸菌科(Acetobacteraceae)、芽孢杆菌科(Bacillaceae)、伯克氏菌科(Burkholderiaceae)、梭菌科(Clostridiaceae)、丛毛单胞菌科(Comamonadaceae)、肠杆菌科(Enterobacteriaceae)、毛螺菌科(Lachnospiraceae)、乳杆菌科(Lactobacillaceae)、甲基杆菌科(Methylobacteriaceae)、莫拉菌科(Moraxellaceae)、鞘脂单胞菌科(Sphingomonadaceae)和草酸杆菌科(Oxalobacteraceae)(Voulgari-Kokotaetal., 2019)。
蜜蜂肠道微生物与宿主多种生命活动息息相关。现有研究表明,肠道微生物在抑制微生物病原的定殖和入侵(Forsgren, 2010; Vásquezetal., 2012; Killeretal., 2014)、复杂碳水化合物的发酵和吸收以及重要营养物质的合成等方面均起到重要作用(Martinsonetal., 2014)。在独栖性野生蜜蜂共生菌群同样有益于宿主的特定功能,Vuong等(2019)研究发现,苜蓿切叶蜂Megachilerotundata共生益生菌米氏乳杆菌Lactobacillusmicheneri对花粉消化、解毒以及病原抑制有一定功效(Vuongetal., 2019);孔蜂Heriadestruncorum共生菌类芽孢杆菌PaenibacillusMBD-MB06具有抑制真菌入侵的作用(Kelleretal., 2018);隧蜂Augochlorapura共生菌中含有大量乳杆菌属Lactobacillus的益生菌群(McFredericketal., 2012)。
社会性蜜蜂的幼虫通过工蜂喂养获得共生微生物,其中肠道共生微生物占有较大比例(Powelletal., 2014)。在刚开始喂养期间,幼虫肠道微生物会迅速增长(Kwong and Moran, 2016),并且,随着幼虫的发育,肠道微生物的群落结构会发生显著变化(Vojvodicetal., 2013)。独栖野生蜜蜂的幼虫在单独的虫室中发育,一般雌蜂将采集到的花粉和花蜜与唾液腺分泌物混合做成蜂粮放入虫室中供幼虫生长发育所需,雌蜂在蜂粮制作的过程定殖必要的有益菌群协助幼虫对蜂粮营养物质的消化(Marthaetal., 1984)。前期研究还发现独栖性野生蜜蜂的肠道微生物具有宿主特异性,并且环境因素可能在独栖性野生蜜蜂蜂巢和宿主共生细菌群落结构和动态变化起着重要驱动作用(Gilliametal., 1990; McFredericketal., 2012, 2017; Kelleretal., 2013)。
大分舌蜂Collelesgigas属膜翅目(Hymenoptera)分舌蜂科(Colletidae)分舌蜂属Colletes,是一种土壤筑巢独栖性野生蜜蜂类群。分舌蜂科是蜜蜂总科中比较原始的一个科,其中分舌蜂属是分布最广的一个属,主要分布于中亚地区(赵延会等, 2010; Niuetal., 2014)。大分舌蜂虫室内有玻璃纸状的膜包裹蜂粮,这种膜具有防水保湿的功能(赵延会等, 2010)。 雌性大分舌蜂主要访问油茶Camelliaoleifera和茶树Camelliasinensis等山茶属植物,是属典型寡食性独栖野生蜜蜂,该蜂成虫期与油茶花期高度吻合,是油茶传粉的优势传粉昆虫(黄敦元等, 2014; Lietal., 2020)。然而,目前为止,还没有关于大分舌蜂肠道微生物的报道(包括成虫和幼虫),鉴于肠道微生物对宿主的重要生物学意义,以及独栖蜜蜂幼虫肠道微生物研究匮乏,本研究采用IlluminaMiSeq二代高通量测序方法,对大分舌蜂幼虫肠道细菌进行了初探,比较了不同龄期幼虫及滞育预蛹肠道细菌群落结构和生物多样性差异,并对肠道细菌的基因功能进行预测。以期为后续此类野生蜜蜂的保护生物学研究提供科学基础和研究思路。
1 材料与方法
1.1 大分舌蜂幼虫及滞育预蛹样品采集
本实验所用大分舌蜂幼虫样本于2018年11月-2019年3月采集于江西省新余市渝水区水北镇(28°04′N, 115°06′E)。参照已有文献报道大分舌蜂巢穴生境(赵延会等, 2010),寻找巢穴进行挖掘与采集,将采集的完好虫室分装于灭菌处理的5 mL冻虫管内并及时带回实验室进行后期处理,剥离大分舌蜂玻璃纸状巢穴,将存活幼虫按照1-5龄和滞育预蛹进行区分(黄敦元等, 2015)。不同龄期幼虫及滞育预蛹的解剖方式:70%酒精进行虫体体表消毒30 s, 0.1%的Triton-X 100溶液处理5 min,最后用超纯水冲洗3次,然后于超净工作台中,在昆虫Ringer’s溶液中解剖得到肠道样品放入2 mL离心管(Limetal., 2015)。累计得到6个实验组,每组5个重复样品,每个样品10头幼虫肠道材料。所有样品经液氮速冻,并及时存入-80℃冰箱内备用。
1.2 大分舌蜂幼虫肠道细菌基因组DNA的提取
肠道细菌基因组DNA提取采用NucleoSpin® 96 Soil Kit(德国MACHEREY-NAGEL公司),提取步骤按试剂盒的说明书。分离得到的DNA用1%琼脂糖凝胶电泳检测并切下目的条带测定,分光光度检测A260/A280和A260/A230值。
1.3 16S rRNA基因片段扩增,文库构建与测序
得到DNA产物利用细菌16S rRNA基因片段V3-V4区通用引物(338F: 5′-ACTCCTACGGGAGG CAGCA-3′; 806R: 5′-GGACTACHVGGGTWTCTAAT-3′)(Lietal., 2018)扩增得到16S rRNA基因片段V3-V4区。结合条形码和参考序列扩增细菌16S rRNA基因V3-V4区。PCR反应体系:DNA样品60 ng, dNTPs(10 μmol/L)1 μL, High GC Enhancer 10 μL, Q5 DNA Polymerase with High-Fidelity 0.2 μL, Buffer 10 μL,上下游引物终浓度10 μmol/L。PCR扩增条件: 95℃预变性5 min; 95℃变性1 min, 50℃退火1 min, 72℃延伸1 min,15个循环; 72℃延伸7 min(Takebayashietal., 2000)。用VAHTSTM DNA Clean Beads纯化第一步得到的PCR产物,随后,在40 μL的反应体系中进行第二轮PCR,反应体系:第一步中PCR产物10 μL, 2×Phμsion HF MM 20 μL, ddH2O 8 μL,上下游引物终浓度10 μmol/L。反应条件: 98℃预变性30 s; 98℃变性10 s, 65℃退火30 s, 72℃延伸30 s, 10个循环; 72℃延伸5 min。最后,用Quant-iTTMdsDNA HS Reagent进行定量,并将其整合到一起。最后,将整合纯化得到的产物利用IlluminaHiseq PE250测序平台进行双端测序。
1.4 测序数据分析
利用FLASH(version 1.2.11)软件对双端数据进行拼接(Magoand Salzberg, 2011);利用Trimmomatic(version 0.33)进行数据过滤过后(Bolgeretal., 2014),除去低于200 bp的序列和大于8 bp的同聚体;利用UCHIME Algorithm去除嵌合体序列,质控获得高质量的clean reads。
利用QIIME V1.7.0(Quantitative Insights Into Microbial Ecology)对同源性在97%水平上进行操作分类单元(operational taxonomic unit, OTU)聚类(Bokulichetal., 2013; Edgar, 2013),并利用SILVA数据库对得到的16S rRNA基因序列进行注释(Quastetal., 2013)。通过RDP(Ribosomal Database Program)(V2.2)在不同分类级别进行分类注释(Wangetal., 2007),并且利用OTUs数据结果进行稀释曲线的绘制,以验证数据的可靠性(Wangetal., 2012)。
基于注释结果,利用Mothur (v.1.30)(Schlossetal., 2009)分析alpha多样性(Shannon-Wiener, Simpson, ACE和Chao1指数);利用SPSS 25.0进行单因素方差分析研究其组间是否存在显著差异;利用GraphPad Prism 8进行可视化。基于QIIME软件聚类得到的OTU,利用Binary-Jaccard算法计算得到组间Beta距离,非度量多维标定法(nonmetric multidimensional scaling, NMDS)降维并进行可视化(Looftetal., 2012);最后,利用多元方差分析PERMANOVA(Adonis)计算组间是否存在显著差异(利用R软件)。利用线性判别分析效应量[linear discriminant analysis (LDA) effect size, LEfSe]揭示出差异显著且最有可能解释细菌群落差异的分类特征(LDA>4.0)(Segataetal., 2011)。基于每个OTU在Greengenes基因数据库(http:∥greengenes.secondgenome.com/)中对应的基因获得COG(Clusters of Orthologous Groups of proteins)蛋白家族信息,利用PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)(Langille-Morganetal., 2013)比对测序数据获得的物种组成信息,推测样本中的功能基因组成,比对测序数据获得的物种组成信息,推测样本中的功能基因组成,利用R软件对数据进行热图绘制,利用SPSS进行单因素方差分析以及多重比较。PICRUSt(http:∥greengenes.secondgenome.com/)和LEfSe(http:∥huttenhower.sph. harvard.edu/lefse/)分析都利用在线Galaxy workflow framework进行。
2 结果
2.1 大分舌蜂幼虫及滞育预蛹肠道细菌16S rRNA基因V3-V4区测序和分类注释
利用IlluminaMiSeq平台对16S rRNA基因V3-V4区扩增子双端测序,共得到2 313 574个原始片段,经过质控优化后得到2 203 686条有效序列(valid reads),平均长度为410.13±3.50 bp(表1)。稀释曲线显示,随着抽样序列数量的增多,每组样本所观测到的物种数趋于平缓饱和,说明本实验的数据具有可靠性(图1)以及反应数据的真实性。根据97%的序列相似度对所有序列进行同源比对,共聚类得到192个OTU (表1),并对结果进行注释得到15个门(注释比例: 99.44%),23个纲(注释比例: 98.87%),43个目(注释比例: 98.87%),80个科(注释比例: 94.92%),128个属(注释比例: 84.75%)(表2)。
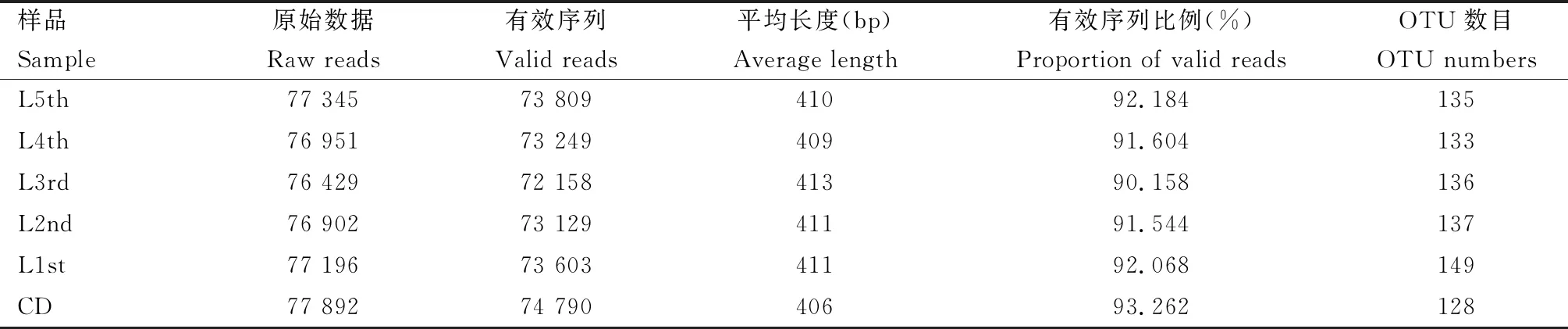
表1 大分舌蜂幼虫及滞育预蛹肠道细菌16S rRNA基因测序后过滤数据
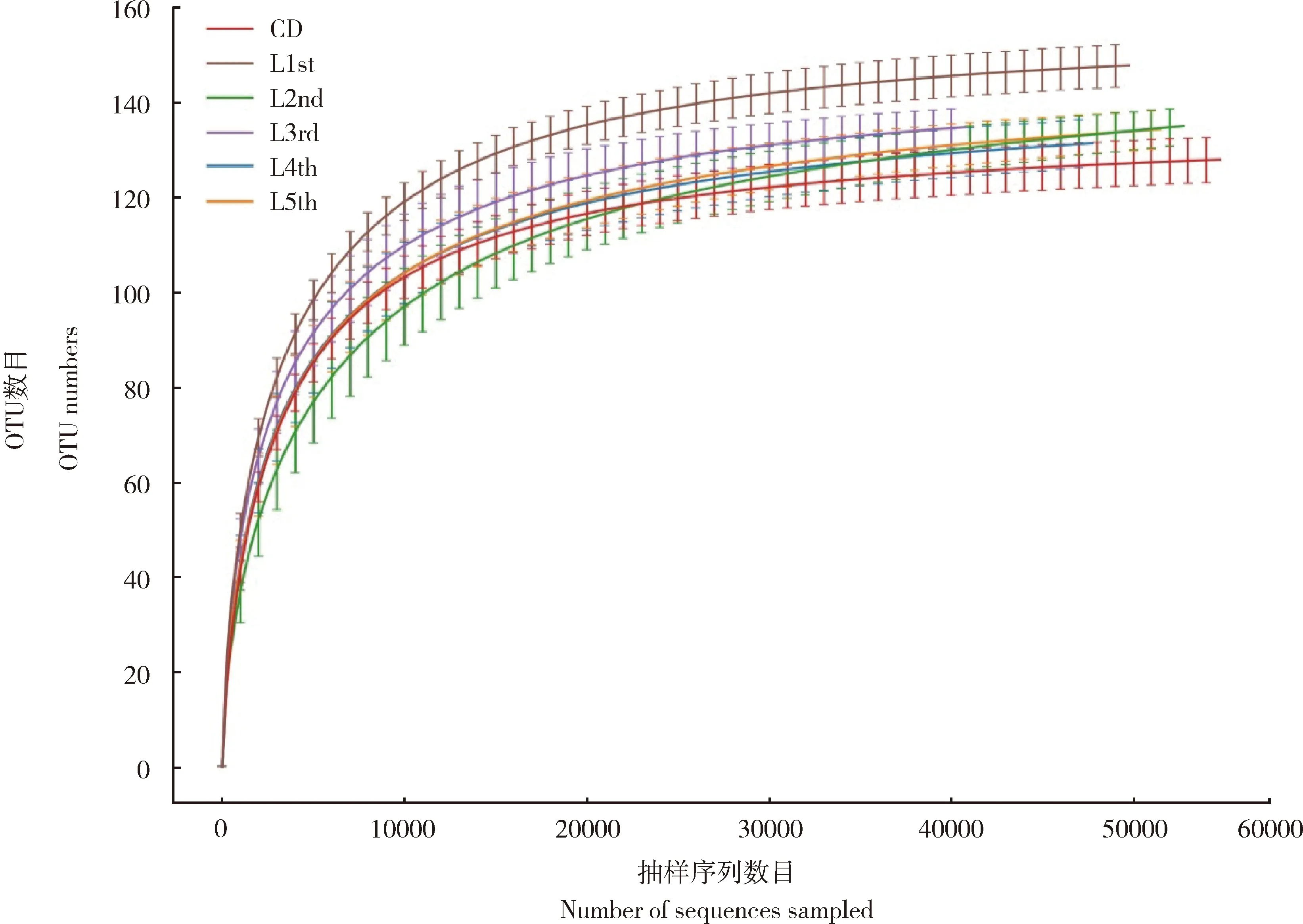
图1 大分舌蜂不同龄期幼虫及滞育预蛹肠道细菌组成稀释曲线
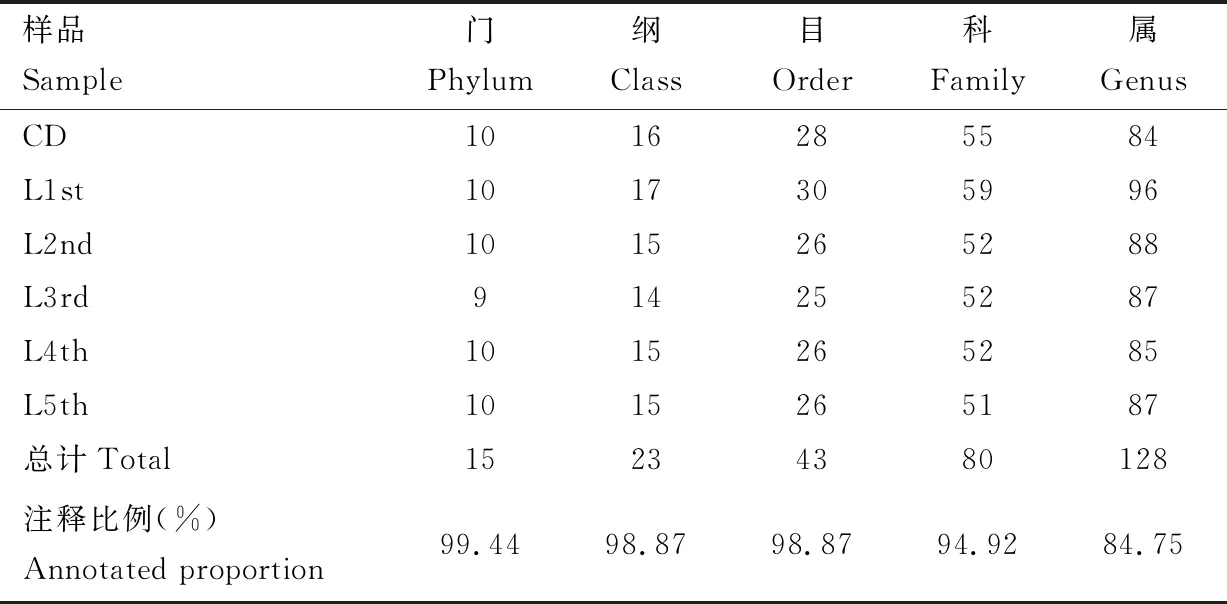
表2 大分舌蜂幼虫及滞育预蛹肠道细菌不同分类级别已注释的分类单元数量统计
2.2 大分舌蜂幼虫及滞育预蛹肠道主要细菌群落组成
从门水平上,主要的10种菌群包括:变形菌门(Proteobacteria)占93.74%(占细菌群落总数的比例;下同),厚壁菌门(Firmicutes)占3.04%,梭杆菌门(Fusobacteria)占1.22%,拟杆菌门(Bacteroidetes)占1.05%, 放线菌门(Actinobacteria)占0.71%,Epsilonbacteraeota占0.05%,酸杆菌门(Acidobacteria)占0.04%,Halanaerobiaeota占0.02%,疣微菌门(Verrucomicrobia)占0.01%,绿弯菌门(Chloroflexi)占0.01%。从目水平,主要的10种菌群包括立克次氏体目(Rickettsiales)占68.68%,肠杆菌目(Enterobacteriales)占19.17%,假单胞菌目(Pseudomonadales)占4.80%,梭菌目(Clostridiales)占1.42%,梭杆菌目(Fusobacteriales)占1.22%,拟杆菌目(Bacteroidales)占0.99%,乳杆菌目(Lactobacillales)占0.75%,根瘤菌目(Rhizobiales)占0.68%,丹毒丝菌目(Erysipelotrichales)占0.43%,棒状菌目(Corynebacteriales)占0.36%。从科水平,主要的10种菌群包括无形体科(Anaplasmataceae)占68.64%,肠杆菌科(Enterobacteriaceae)占19.17%,莫拉氏菌科(Moraxellaceae)占4.78%,梭杆菌科(Fusobacteriaceae)占1.22%,毛螺菌科(Lachnospiraceae)占0.66%,根瘤菌科(Rhizobiaceae)占0.62%,普雷沃氏菌科(Prevotellaceae)占0.53%,乳杆菌科(Lactobacillaceae)占0.49%,消化链球菌科(Peptostreptococcaceae)占0.44%,韦荣球菌科(Erysipelotrichaceae)占0.43%。从属水平,主要的10种菌群包括:沃尔巴克氏体属Wolbachia占68.64%,肠杆菌属Enterobacter占18.18%,不动杆菌属Acinetobacter占4.77%,梭杆菌属Fusobacterium占1.22%,苍白杆菌属Ochrobactrum占0.61%,乳杆菌属Lactobacillus占0.49%,拟杆菌属Bacteroides占0.40%,Peptoclostridium占0.36%,泛菌属Pantoea占0.36%,红球菌属Rhodococcus占0.35%。物种组成结果显示,低龄组(1-3龄幼虫)肠道细菌组成上相近,高龄组(4-5龄幼虫)肠道细菌组成上相近,滞育预蛹肠道细菌组成上与其他龄期幼虫的差异较大(图2)。
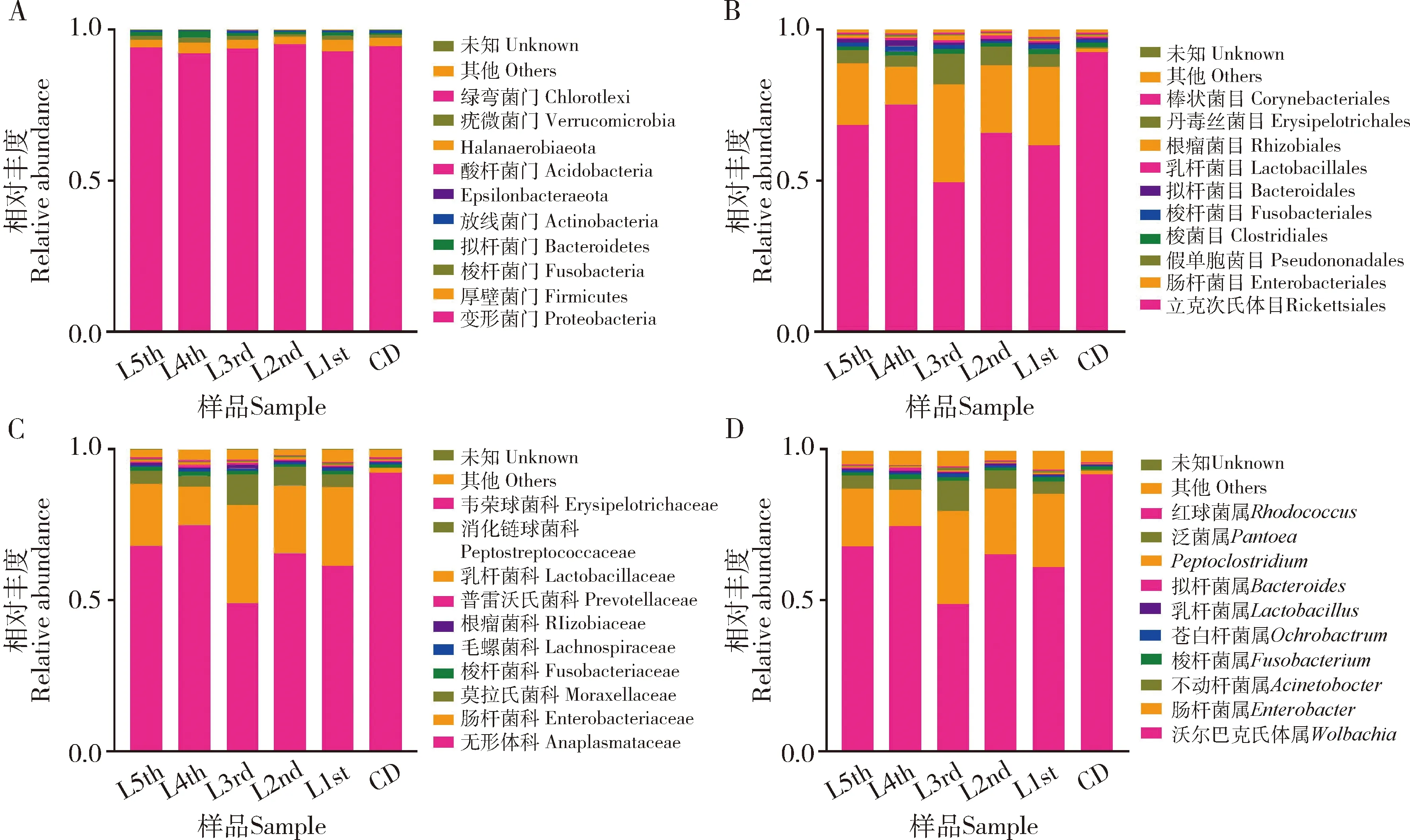
图2 大分舌蜂幼虫及滞育预蛹肠道细菌在门(A)、目(B)、科(C)和属(D)水平的相对丰度
2.3 大分舌蜂幼虫及滞育预蛹肠道细菌群落多样性
基于属水平Alpha多样性分析结果表明(图3):ACE与Chao1指数在低龄组(1-3龄幼虫)、高龄组(4-5龄幼虫)与滞育预蛹组之间不存在显著差异。低龄组的Shannon-Wiener指数极显著高于滞育预蛹组(P<0.001),高龄组的Shannon-Wiener指数极显著高于滞育预蛹组(P<0.01),高龄组与低龄组Shannon-Wiener指数没有显著差异,总体上,低龄组到滞育预蛹组出现Shannon-Wiener指数从高到低的趋势。低龄组的Simpson指数极显著低于滞育预蛹组(P<0.001),高龄组的Simpson指数极显著低于滞育预蛹组(P<0.01),高龄组与低龄组的Simpson指数没有显著差异(P>0.05),总体上,低龄组到滞育预蛹组,出现Simpson指数从低到高的趋势。
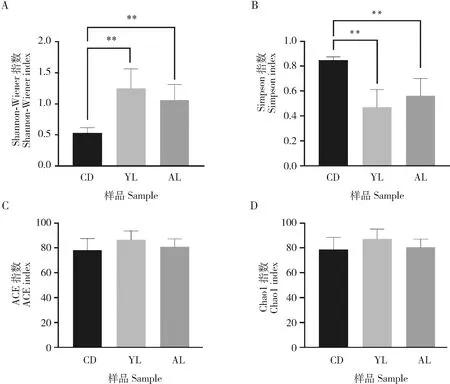
图3 大分舌蜂幼虫及滞育预蛹肠道细菌α多样性指数差异分析
Beta多样性分析结果表明,1-3龄幼虫肠道细菌聚在一起,4和5龄幼虫肠道细菌聚在一起,滞育预蛹期肠道细菌聚集在一起,显示出低龄组、高龄组和滞育预蛹组3个水平(图4);进一步对其组间距离进行多元方差分析,结果显示低龄组(YL)与高龄组(AL)肠道细菌群落结构存在显著差异(R2=0.080,P=0.008),高龄组(AL)与滞育预蛹组(CD)肠道细菌群落结构比较有显著差异(R2=0.310,P=0.001),滞育预蛹组(CD)和低龄组(YL)肠道细菌群落结构比较有显著差异(R2=0.291,P=0.001),显示大分舌蜂幼虫肠道细菌群落结构变化调节过程可分为低龄组、高龄组和滞育预蛹组。
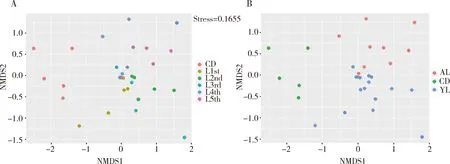
图4 利用非度量多维尺度法(NMDS)对大分舌蜂幼虫及滞育预蛹肠道细菌进行Beta多样性分析
2.4 大分舌蜂不同阶段幼虫及滞育预蛹肠道菌群差异
线性判别分析(LDA)效应量(LEfSe)分析结果显示(图5),滞育预蛹组(CD)与低龄组(YL)都存在显著优势的菌群,而高龄组(AL)不存在显著优势菌群。γ-变形菌纲(Gammaproteobacteria)为低龄组优势纲,α-变形菌纲(Alphaproteobacteria)为滞育预蛹组优势纲;在目水平,肠杆菌目(Enterobacteriales)和假单胞菌目(Pseudomonadales)为低龄组优势目,立克次氏体目(Rickettsiales)为滞育预蛹组优势目;在科水平,肠杆菌科(Enterobacteriaceae)和莫拉菌科(Moraxellaceae)为低龄组优势科,无形体科(Anaplasmataceae)为滞育预蛹组优势科;在属水平,肠杆菌属Enterobacter和不动杆菌属Acinetobacter为低龄组优势属,沃尔巴克氏菌属Wolbachia为滞育预蛹组优势属。
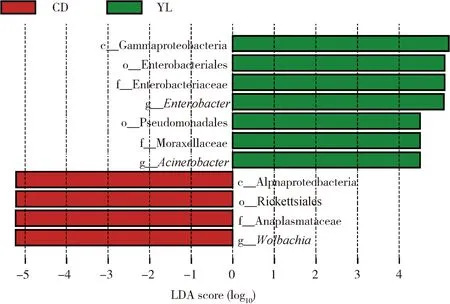
图5 大分舌蜂幼虫及滞育预蛹肠道细菌LEfSe分析
2.5 大分舌蜂幼虫及滞育预蛹肠道菌群基因功能预测
利用COG数据库对大分舌蜂幼虫肠道菌群进行基因功能预测,进一步聚类分析发现,低龄组(YL)和高龄组(AL)聚在一个分支而滞育预蛹组(CD)为单独一分支(图6)。同源族分析结果显示,注释到的功能包括三大类:信息储存与处理(information storage and processing)(占25.02%), 新陈代谢(metabolism)(占38.77%),细胞生理过程与信号(cellular physiological processes and signaling)(占21.02%)。低龄组新陈代谢和细胞生理过程与信号相关功能基因比例显著高于滞育预蛹组的(P<0.05),滞育预蛹组信息储存与处理相关功能基因比例显著高于低龄组(P<0.01)和高龄组的(P<0.05)(图7)。进一步分析发现,滞育预蛹组肠道菌群在细胞生理过程与信号相关功能基因大类中的细胞内转运、分泌和囊泡运输(intracellular trafficking, secretion and vesicular transport),翻译后修饰、蛋白转换、伴侣(posttranslational modification, protein turnover, chaperones)和细胞周期控制、细胞分裂、染色体分配(cell cycle control, cell division, chromosome partitioning)相关基因显著高于低龄组与高龄组的。滞育预蛹组肠道菌群在信息储存与处理相关功能基因大类中的复制、重组和修复(replication, recombination and repair)以及翻译、核糖体结构和生物发生(translation, ribosomal structure and biogenesis)相关基因显著高于低龄组与高龄组的。
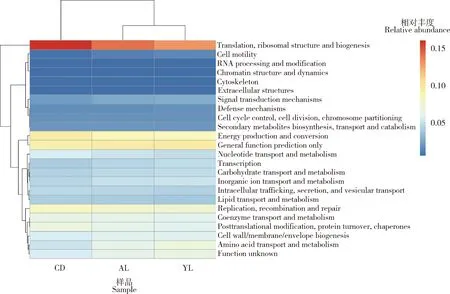
图6 大分舌蜂幼虫及滞育预蛹肠道细菌基因功能COG注释及聚类分析
3 讨论
本研究发现了大分舌蜂幼虫肠道细菌与之前独栖蜜蜂肠道核心菌群(Voulgari-Kokotaetal., 2019)相似的一些共生菌群,例如肠杆菌科、莫拉菌科、毛螺菌科和乳杆菌科。大分舌蜂幼虫肠道细菌群落中,变形菌门占据最主要地位,这个门的细菌在社会性蜜蜂中也比较常见。在A.melliferajemenitica中,肠道细菌主要的3个门为变形菌门、厚壁菌门和放线菌门(Andersonetal., 2015);另外在日本东方蜜蜂Apisceranajaponica共生细菌研究中也发现变形菌门的存在(McFredericketal., 2017)。但本研究厚壁菌门只占整个菌群的很小比例。另外,在属分类水平上,沃尔巴克氏菌Wolbachia是大分舌蜂幼虫肠道共生细菌中的最主要的属,同时,本研究也发现了一些独有的肠道共生细菌,这些细菌的存在可能与宿主特异性,以及环境食物来源相关。大分舌蜂具有典型的寡食性,其主要访花山茶属的植物(油茶占主要地位),虫室具有玻璃纸状的独特结构与土壤筑巢的特点(赵延会等, 2010; 黄敦元等, 2015),肠道共生菌群可能受到山茶属植物与土壤共生菌群的影响,然而,其中是否具有相关性以及影响程度还有待通过其他实验进行验证。
Beta多样性分析结果显示,大分舌蜂在低龄幼虫(1-3龄)、高龄幼虫(4-5龄)和滞育预蛹不同阶段肠道共生细菌的组成结构出现了显著变化;Alpha多样性分析显示,随着幼虫生长阶段的递进,其肠道微生物的多样性出现逐步降低的现象(图3)。相反,群居社会性蜜蜂幼虫从低龄到高龄的发育过程中,其肠道细菌多样性表现出逐步升高的趋势(Kwong and Moran, 2016),这一现象可能与幼虫生长过程中进食息息相关。前期的行为学研究显示,大分舌蜂低龄幼虫期(1-3龄)进食相对缓慢,高龄幼虫期(4-5龄)开始大量进食,直到食物耗尽停止进食,便进入滞育预蛹,这一点可能与其进食特点具有相关性。另一方面,大分舌蜂整个幼虫期的食物都来源于成蜂提前备好的蜂粮,并且整个周期都存活于相对封闭、稳定的玻璃纸状巢室中(黄敦元等, 2015),这与群居蜜蜂幼虫通过成蜂喂养摄取食物的口器喂养行为有明显的不同,因此,其变化趋势或许也与肠道微生物驱动对相对稳定环境的适应有关。前期研究表明,动物肠道菌群对不同类型食物胁迫下引起的肠道环境变化存在逐渐适应的特征(Tiloccaetal., 2017)。但这种随着龄期增加,其肠道细菌多样性降低的现象以及其如何对肠道环境逐步适应这一过程还有待进一步研究。
本研究进一步对大分舌蜂低龄幼虫、高龄幼虫和滞育预蛹3个阶段肠道细菌之间的差异进行分析,以找到不同阶段出现显著变化的物种,这些物种的显著变化与存在可能为不同阶段大分舌蜂幼虫宿主提供着重要的生物学意义。沃尔巴克氏菌属是本研究结果中占比最多的一个属,并且,它在滞育预蛹组占比更为显著。作为一种广泛存在于昆虫体内的共生菌群,其功能是多样的。沃尔巴克氏体的一个关键特征是它能在无脊椎动物体内生存并操纵细胞和生殖过程(Werrenetal., 2008)。其表型效应和宿主相互作用主要表现为:一是对精子产生修饰作用从而导致精卵细胞胞质不亲和(cytoplasmic incompatibility, CI),最后造成胚胎的早期死亡(Poinsotetal., 2003);其次是通过多种方式引起宿主孤雌生殖,例如:作用于细胞减数第一次分裂,导致中期染色体未分离而造成纯合二倍体核的形成,诱导孤雌生殖(parthenogensis induction)(Weeks and Breeuwer, 2001);三是通过在雄性激素腺体内增殖而影响其功能造成宿主雌性化(feminization)(Vandekerckhoveetal., 2003)以及影响宿主繁殖力(周淑香等, 2009)等多种作用。另外,研究发现,沃尔巴克氏菌属在温带臭虫Cimexlectularius生存和繁衍中是必不可少的,它在宿主中起着营养互惠(nutritional mutualism)的作用(Hosokawaetal., 2010);此外,沃尔巴克氏菌属在抑制病原中也存在有利作用,例如可以诱导宿主对病原产生抗性,并且之前的研究显示,其显著降低RNA病毒感染造成的黑腹果蝇Drosophilamelanogaster死亡(Hedgesetal., 2008; Teixeiraetal., 2008)。Walker等(2011)的研究显示,沃尔巴克氏菌属可以阻断登革热2型(DENV-2)在埃及伊蚊Aedesaegypti中的传播。在低龄组(1-3龄幼虫)有两个属显示出优势,包括肠杆菌属Enterobacter和不动杆菌属Acinetobacter(图5)。 尽管,目前对于肠杆菌属Enterobacter的研究中,未表明它是一种益生菌群(Al-Ghamdietal., 2017),但这一菌群却在蜜蜂肠道中有所发现(Martinsonetal., 2011),并且在A.melliferajemenitica中被分离得到(Khanetal., 2017)。不动杆菌属Acinetobacter由于具有多样性的代谢功能而广泛存在于各种环境中,同样也在蜜蜂肠道中被分离得到过,例如蜂属不动杆菌A.apis和蜜源不动杆菌A.nectaris(Kimetal., 2014),其中蜜源不动杆菌是一种常见花和蜜蜂的共生微生物(álvarez-Pérezetal., 2013)。然而,目前为止,这一细菌群的成员对寄主的有利功能还未见报道,因此仍有待研究。上述结果表明,大分舌蜂幼虫不同发育阶段存在不同的优势菌群,其可能在不同发育阶段发挥着重要作用,但其中具有特定功能的菌株还有待进一步分析以及实验证明。
通过PICRUSt对低龄组、高龄组以及滞育预蛹组肠道共生菌基因功能COG预测。聚类热图显示,低龄组与高龄组聚在一个分支,而滞育预蛹组单独为一个分支(图6)。表明低龄组与高龄组幼虫肠道细菌基因功能更为相近,而滞育预蛹组与发育阶段肠道细菌(低龄组与高龄组)在基因功能上较远。这也进一步从基因功能角度揭示大分舌蜂幼虫及滞育预蛹具有的3个发育阶段(低龄幼虫、高龄幼虫以及滞育预蛹阶段)的特点(黄敦元等, 2015)。同时,研究发现,低龄组新陈代谢和细胞生理过程与信号相关功能基因比例显著高于滞育预蛹组;滞育预蛹组信息储存与处理相关功能基因比例显著高于低龄和高龄组(图7)。低龄与高龄幼虫都处于不断进食的生长期,对于刚开始进食的低龄幼虫来讲,新陈代谢与细胞生理过程与处理相关功能基因的显著存在有助于宿主对食物营养利用以及宿主的生长;而滞育预蛹处于停止进食并且表现为形态变化停顿的时期,信号储存与处理相关功能基因的显著存在有助于宿主在没有食物来源的情况下维持存活。进一步分析显示,滞育预蛹期肠道菌群在细胞内转运、分泌和囊泡运输,翻译后修饰、蛋白转换、伴侣蛋白,细胞周期控制、细胞分裂、染色体分配,复制、重组和修复以及翻译、核糖体结构和生物发生等相关功能基因都显著高于低龄组与高龄组。综上,结果表明,在大分舌蜂幼虫不同发育阶段中,其肠道菌群可能发挥着不同功能。然而,基于16S rRNA基因片段的PICRUSt分析并不能完全说明其中真正存在的功能,因此,这些功能是否与大分舌蜂幼虫的生长发育存在直接相关性以及肠道细菌是否在其生长发育中发挥着具体作用还有待通过宏基因组学或其他技术手段进一步研究。
猜你喜欢
预防青少年犯罪研究(2022年2期)2022-08-17 06:26:42
家庭医学(下半月)(2020年3期)2020-05-30 12:42:04
心肺血管病杂志(2019年6期)2019-07-12 09:04:30
金色少年(奇趣科普)(2017年4期)2017-06-05 15:03:48
农村农业农民·B版(2016年7期)2016-10-21 10:44:23
西南军医(2016年2期)2016-01-23 02:14:10
实用手外科杂志(2015年4期)2015-08-27 01:54:10
中国当代医药(2015年24期)2015-03-01 02:06:21
西南军医(2015年1期)2015-01-22 09:08:36
天然产物研究与开发(2014年8期)2014-04-27 14:16:30
