
锦鲤肠道不同部位菌群组成结构及多样性分析
2021-06-30 03:14孙中石范艳蕊吕爱军胡秀彩石洪玥陈丽梅孙敬锋
南方农业学报 2021年2期
孙中石 范艳蕊 吕爱军 胡秀彩 石洪玥 陈丽梅 孙敬锋


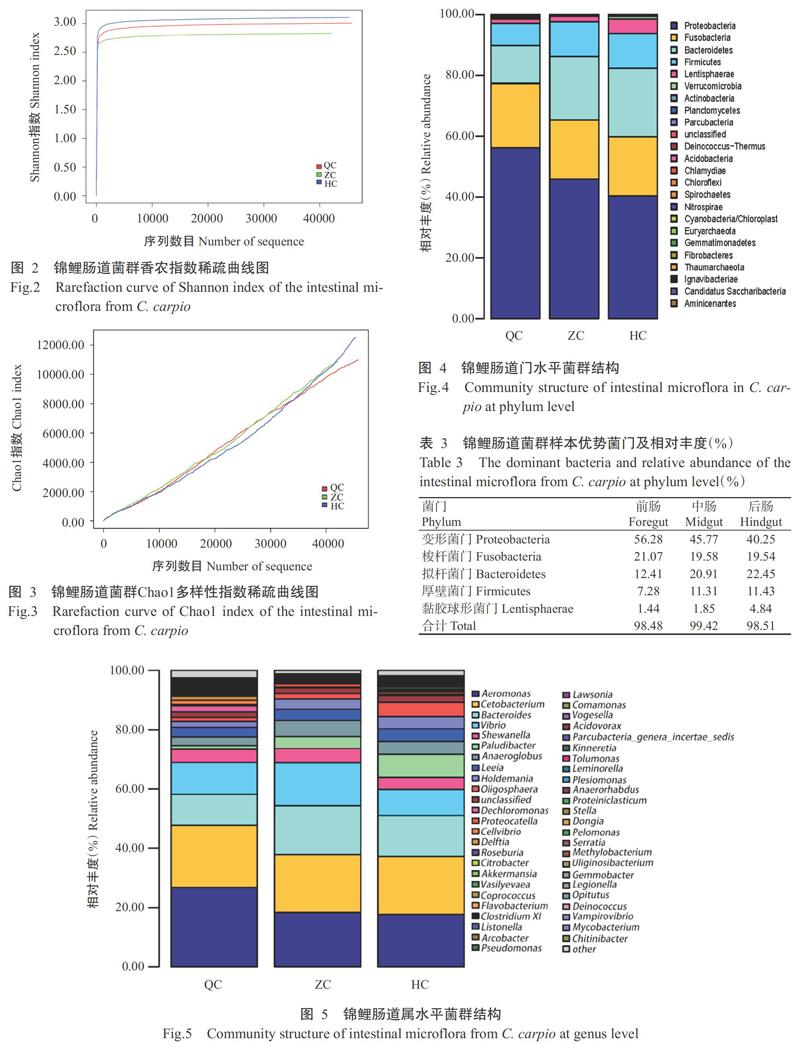
摘要:【目的】研究錦鲤(Cyprinus carpio var. koi)肠道的菌群组成结构及多样性,为深入研究锦鲤肠道菌群的功能及肠道不同部位的生理功能的分化提供参考。【方法】收集锦鲤前肠、中肠和后肠的肠道细菌,通过传统培养技术,采用细菌微量生化鉴定管对肠道细菌进行各项生理生化指标的测定;分别提取3个肠段的肠道菌群基因组DNA,进一步采用16S rDNA高通量测序技术,分析3个肠段的菌群组成和生物多样性。【结果】前肠、中肠和后肠样本测序后分别得到48659、47013和47819条有效序列,按97%相似性水平划分OTU后,分别得到1555、1294和1423个OUTs。α多样性分析结果显示,后肠菌群样本的Shannon指数、Chao1指数最高。在门水平上,前肠、中肠和后肠丰度最高的5个菌门均是变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和黏胶球形菌门(Lentisphaerae),推测这些菌门是锦鲤肠道的核心细菌类群,但各菌门在不同部位的相对丰度有差异。构建的物种丰度热图显示相对丰度最高的前10类菌属相同,但某些菌属在肠道不同部位的相对丰度有较大差异,如代尔夫特菌属(Delft)、弓形菌属(Toxoplasma)。UniFrac分析显示中肠和后肠的肠道菌群聚为一支,表明这2个部位菌群组成结构相似度较高。【结论】锦鲤肠道不同部位具有独特的菌群结构,可能会影响肠道不同部位功能的分化。
关键词: 锦鲤;肠道菌群;多样性;16S rDNA
中图分类号: S917.4 文献标志码: A 文章编号:2095-1191(2021)02-0483-08
Abstract:【Objective】This study aimed to investigate the diversity and community of microflora from the three intestinal segments(foregut,midgut,and hindgut) of koi carp(Cyprinus carpio var. koi) and provide reference for further research on the function of koi carpintestinal flora and the differentiation of physiological functions in different parts of the intestine. 【Method】Microflora from the three intestinal segments, foregut,midgut,and hindgut, were collected, and the physiological and biochemical indexes of intestinal bacteria through the bacterial microbiochemical identification tube were identified based on the traditional bacterial culture technique. Genomic DNA of intestinal microflora from the three intestinal segments was extracted,and 16S rDNA high-throughput sequencing technology was used to analyze the microflora composition and diversity. 【Result】The three microflora samples from the three intestinal segments(foregut,midgut,and hindgut) were sequenced to obtain 48659,47013,47819 effective tags, respectively. After dividing the OTUs based on 97% sequence similarity level,1555,1294, and 1423 OUTs were obtained, respectively. Alpha diversity analysis showed that the Shannon and Chao1 indexes of the hindgut flora samples were the highest. At the phylum level, the top five phyla with the highest abundance in the foregut,midgut,and hindgut were all Proteobacteria,Fusobacteria,Bacteroidetes,Firmicutes and Lentisphaerae,which could be probably recognized as the core bacterial taxa in the koi carpintestine, but the relative abundance of each phylum was different in different parts. The species abundance heat map constructed showed that the top ten species with the highest relative abundance were the same, but the relative abundance of some species in different parts of the intestine was quite different, such as Delft and Toxoplasma. The UniFrac cluster heat map analysis showed that the intestinal flora of the midgut and hindgut were clustered into one group, indicating that the composition of the flora of these two parts was highly similar in structure. 【Conclusion】Unique microflora structures are formed in different parts of the koi carp intestinal tract, and may affect the functional differentiation of different parts of the intestinal tract.
Key words: koi carp(Cyprinus carpio var. koi); intestinal microflora; diversity; 16S rDNA
Foundation item: National Natural Science Foundation of China(31972840); Tianjin Natural Science Foundation (19JCZDJC34600); Undergraduate Innovation and Entrepreneurship Training Program of Tianjin(202010061061)
0 引言
【研究意义】肠道是动物消化吸收营养物质的主要场所,同时也参与机体的免疫防御过程。鱼类肠道是一个十分复杂的生态系统,肠道内生存着大量的微生物,其功能包括抵抗细菌感染、分解营养物质以及为宿主提供酶、氨基酸和维生素等生命活动所必需物质(Sugita et al.,1997)。因此,鱼类肠道微生物与机体健康及生理状况有着密切的关系(Ganguly and Prasad,2012)。肠道微生物组成结构的变化能够引起宿主生理状态及免疫机能的改变,其微生态系统紊乱会增加宿主的患病风险(Nayak,2010;Brown et al.,2012),许多研究者使用益生菌来维持或改善鱼类肠道菌群的组成结构,增强机体抗病力及抗应激能力(Verschuere et al.,2000)。因此,研究鱼类肠道菌群的组成结构和多样性是了解其与机体生理功能间相互关系的重要基础。【前人研究进展】动物肠道微生物的研究方法总体上经历了3个阶段,分别是依赖于人工培养的生理生化鉴定方法、传统分子生物学方法及基于高通量测序的宏基因组学方法(许文涛等,2009)。依赖于细菌人工培养的生理生化鉴定方法成本低廉,但在培养过程中存在菌种比例发生变化、很多细菌无法培养等缺陷,这些因素导致无法准确了解肠道菌群的组成结构特征。传统分子生物学方法主要有实时定量PCR(qPCR)和变性梯度凝胶电泳(DGGE)。应用qPCR方法每次只能鉴定一类微生物,存在通量低的缺陷;DGGE方法因受限于其较低的灵敏度,只能应用于肠道中较高丰度微生物的鉴定(李东萍等,2015)。基于16S rDNA高通量测序的细菌鉴定方法将16S rDNA在菌种鉴定方面的优势与高通量测序技术的特点相结合,实现了对肠道微生物的快速准确鉴定和定量(李东萍等,2015)。目前一些鱼类肠道的菌群组成及多样性已被报道,如罗非鱼(Oreochromis mossambicus)(Giatsis et al.,2015)、黑头软口鲦(Pimephales promelas)(Narrowe et al.,2015)、锦鲤(Cyprinus carpio var. koi)(Han et al.,2018)。然而,高等动物肠道不同部位通常承担不同的生理功能。在小鼠、猪和鸡等高等动物中(Gu et al.,2013;Looft et al.,2014;Choi et al.,2015)已报道了不同肠段中微生物群组成及多样性的差异。在鱼类低等脊椎动物的研究中也证明肠道不同部位各种消化酶、水解酶、氧化酶等具有差异分布,揭示了鱼类肠道不同部位在消化吸收、免疫防御功能上的分化(Wang et al.,2017)。【本研究切入点】目前在鱼类肠道菌群的研究中,通常将整个肠道的菌群进行鉴定分析,尚未见涉及肠道不同部位的菌群结构差异,而研究鱼类不同肠段微生物组成及多样性,能够更深入地了解鱼类肠道菌群的功能和作用。【拟解决的关键问题】锦鲤(Cyprinus carpio var. koi)是主要养殖观赏鱼种类之一,具有较高的观赏价值。本研究利用16S rDNA高通量测序技术,研究锦鲤肠道3个肠段(前肠、中肠和后肠)的菌群组成及多样性,分析其菌群组成結构的差异,并应用基于传统培养技术的生理生化试验方法对3个肠段的细菌进行鉴定,以期为深入研究锦鲤肠道菌群的功能及肠道不同部位的生理功能分化提供参考。
1 材料与方法
1. 1 试验材料
试验所用健康锦鲤购自天津功旺锦鲤养殖基地,平均体长为32.60±1.66 cm,平均体重为398.90±19.71 g。锦鲤在实验室暂养7 d后用于试验,养殖期间水温控制在20 ℃左右,连续曝气充氧,每日饲喂2次鱼体重1%的普通商业饲料。
1. 2 肠道菌群样品制备
取5尾锦鲤进行麻醉,以70%的酒精棉球擦拭鱼体表,并用无菌手术剪将肠道从腹腔中分离,按照解剖学特征将其分为3段,分别是前肠、中肠和后肠(Wallace et al.,2005)。将3个肠段的内容物分别挤出后收集,然后剖开肠道,刮取肠粘膜。将5尾鱼3段肠道分离出的内容物及肠黏膜分别放入3个50 mL离心管,混匀,-80 ℃保存备用。前、中、后3段肠道的菌群样本分别命名为QC、ZC、HC。
1. 3 基于培养技术的生理生化方法鉴定肠道细菌
1. 3. 1 细菌培养 从前肠、中肠和后肠的肠道样本中各取0.1 g用于肠道细菌培养。用无菌生理盐水对0.1 g样品进行倍比稀释,得到各梯度稀释细菌悬液(10-1、10-2、10-3、10-4、10-5、10-6)。分别取100 ?L细菌悬液涂布于LB固体培养基,每个稀释度涂布3个平板,随后在28 ℃培养箱倒置培养18 h。选取菌落清晰、分散良好且菌落数为30~100的培养平板进行菌落计数及形态观察。在挑选的培养平板中,随机选取前肠、中肠和后肠样本的各50个菌落作为待测菌株,依次编号,进行增殖培养,纯化后的菌液与15%甘油1∶1混合后置于-20 ℃保存备用。
1. 3. 2 生理生化试验 生理生化试验鉴定项目包括革兰氏染色、细菌动力试验、细菌氧化酶试验、细菌氧化发酵试验、细菌需钠试验等。参照《一般细菌常用鉴定方法》(中国科学院微生物研究所细菌分类组1978)和《伯杰氏细菌鉴定手册 第9版》(英文版)(Garrity 2005)的方法,在属水平上对各菌株进行分类鉴定。
1. 4 16S rDNA高通量测序方法鉴定肠道细菌
1. 4. 1 肠道菌群基因组DNA的提取 取剩余的前肠、中肠和后肠的肠道菌群样品,参照Han等(2018)的方法进行肠道微生物基因组DNA提取,利用1%琼脂糖凝胶电泳进行检测,然后送至生工生物工程(上海)股份有限公司进行16S rDNA高通量测序。
1. 4. 2 16S rDNA高通量测序 设计针对细菌16S rDNA V3~V4区的引物进行PCR扩增,上游和下游引物:341F(5'-CCCTACACGACGCTCTTCCGATCTG CCTACGGGNGGCWGCAG-3');805R(5'-GACTG GAGTTCCTTGGCACCCGAGAATTCCAGACTA CHVGGGTATCTAATCC-3')。PCR反应体系50.0 μL:10×PCR Buffer 5.0 μL、dNTP(10 mmol/L)0.5 μL、DNA 10 ng、Bar-PCR primer F(50 μmol/L)0.5 μL、Primer R(50 μmol/L)0.5 μL、Plantium Taq(5 U/μL)0.5 μL,加ddH2O补足至50.0 μL。反应程序:94 ℃预变性3 min;5次循环(94 ℃ 30 s,45 ℃20 s,65 ℃ 30 s),20次循环(94 ℃ 20 s,55 ℃ 20 s,72 ℃ 30 s);72 ℃延伸5 min。用SanPrep柱式DNA凝胶提取试剂盒(Biotech,美国)纯化PCR扩增产物。最后,利用Illumina MiSeq平台对PCR产物进行测序。
1. 5 数据分析
采用Cutadapt、Pear、Prinseq软件处理原始数据。首先去除引物接头序列,将成对的核苷酸单链拼接成一条序列,然后按照标签序列识别区分不同样品,得到各样本数据,最后对各样本数据的质量进行质控过滤,得到各样本的有效数据。将得到的序列在97%的相似水平下聚类成操作分类单元(OTUs)。根据OTU分类计算样本的Chao1指数和Shannon指数。将得到的OTU序列利用BLASTn与对应数据库进行比对,筛选出OTU序列的最佳比对结果,将样本中物种丰度前50以外的物种全部合并为other。将有效序列进行系谱分类,并在不同分类水平统计肠道菌群优势细菌类群及其相对丰度。最后,基于UniFrac分析通过各样本基因序列间的进化信息比较样本在特定的进化谱系中是否有显著的微生物群落差异。
2 结果与分析
2. 1 基于培养技术的生理生化试验鉴定结果
接种稀释菌液培养18 h,根据出现的菌落数计算得出,前肠、中肠和后肠的细菌含量分别为8.28×106、2.91×106和2.68×106 CFU/g。通过生理生化试验在属水平上3个肠段的菌群共鉴定到9个类群,其中气单胞菌属(Aeromonas)、肠杆菌属(Enterobacter)和弧菌属(Vibrio)在3段肠道中均为优势菌。前肠丰度最高的类群为肠杆菌属(24%),中肠丰度最高的类群为气单胞菌属(58%),后肠丰度最高的类群为弧菌属(52%)。除此之外,还检测出不动杆菌属(Acinetobacter)、黄杆菌属(Flavobacterium)、假单胞菌属(Pseudomonas)、产碱菌属(Alcaligenes)、棒杆菌属(Corynebacterium)及葡萄球菌属(Staphylococcus)(表1)。
2. 2 16S rDNA高通量测序分析
2. 2. 1 菌群OTU聚类及多样性分析 由前肠、中肠和后肠的3个样本分别得到48659、47013和47819条原始序列,经过质量控制、除去嵌合体与非靶区域序列后,得到的序列数目分别为47673、46465和47099(表2)。以97%相似性水平为标准划分OTU,所有序列被分为3698个OTUs(图1),其中,前肠、中肠和后肠样品的OTU数分别是1555、1294和1423,所占比例分别为42.05%、34.99%和38.48%。α多样性分析结果显示,3个肠段菌群共有的OTUs为196个,占总数的5.30%。前肠、中肠和后肠菌群样品的香农指数分别为3.01、2.83和3.11(图2),Chao1指数分别为10983.61、10881.23和12519.16(图3)。
2. 2. 2 菌群组成结构分析 在门分类水平上,前肠、中肠和后肠丰度最高的5个菌门依次是变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和黏胶球形菌门(Lentisphaerae)(图4)。上述5个菌门在前肠中依次占其菌群总数的56.28%、21.07%、12.41%、7.28%和1.44%,在中腸依次占其菌群总数的45.77%、19.58%、20.91%、11.31%和1.85%,在后肠中依次占其菌群总数的40.25%、19.54%、22.45%、11.43%和4.84%(表3)。
在属分类水平上,前肠、中肠和后肠丰度最高的前10个类群依次是气单胞菌属(Aeromonas)、鲸杆菌属(Cetobacterium)、拟杆菌属(Bacteroides)、弧菌属(Vibrio)、希瓦氏菌属(Shewanella)、沼杆菌属(Paludibacter)、厌氧球形菌属(Anaeroglobus)、Leeia、霍尔德曼氏菌属(Holdemania)和寡球菌属(Oligosphaera)(图5);丰度最高的前10个菌属在前肠、中肠和后肠样品中分别占菌群总数的84.23%、92.23%和89.28%(表4)。在不同肠段中还存在丰度差异的其他菌属,如代尔夫特菌属(Delftia)和弓形杆菌属(Arcobacter)在前肠中的丰度较高,梭状芽孢杆菌(Clostridium)在前肠中的丰度较低,Lawsonia在后肠中的丰度较高(图6)。
2. 2. 3 相似性分析 通过UniFrac分析构建聚类热图展现样本间的距离关系。如图7所示,中肠与后肠的肠道菌群聚为一支,表明二者菌群结构相似度较高;前肠与中肠、后肠菌群结构差异较大。
3 讨论
动物肠道菌群的形成受生长环境、饲料营养成分及遗传等因素的多重影响(Ley et al.,2006),菌群组成结构是外界环境、宿主肠道内环境和微生物之间选择和共同进化的结果(Ley et al.,2008)。本研究中,α多样性分析显示锦鲤后肠的微生物多样性最高,前肠次之,中肠最低。在不同的分类水平下,前肠、中肠和后肠大部分细菌类群的丰度存在差异。汤伏生等(1994)曾报道,普通鲤鱼前肠细菌大部分靠吸收宿主营养而生存,中肠和后肠细菌主要靠吸收食物中的养分而生存,表明中肠和后肠细菌在营养获取方式上与前肠细菌具有明显区别。本研究中,锦鲤前肠与中肠和后肠的菌群组成结构差异性较大,中肠与后肠的菌群组成结构相似度更高。该现象可能与细菌营养获取方式不同有关,另外,很多鱼类前肠具有特异的解剖学特征(Wallace et al.,2005),这些特征使前肠的细菌谱系获得不同的选择压力(Lan and Love,2012),从而形成了较为独特的菌群结构。
关于陆生哺乳动物肠道菌群的研究表明,拟杆菌门和厚壁菌门是最主要的优势细菌门类(Bowman and McCuaig,2003;Qin et al.,2010)。在本研究结果中,锦鲤肠道中丰度最高的是变形菌门,其次是梭杆菌门。据报道,变形菌门细菌种类在水环境及海底沉积物等环境中广泛存在(Ley et al.,2008)。鱼类肠道微生物易受到外界养殖环境的影响(Tsuchiya et al.,2008),可能是变形菌门成为锦鲤肠道优势菌群的重要原因。不仅在锦鲤肠道,黄颡鱼、斑马鱼和草鱼肠道中也普遍存在变形菌门细菌类群(Han et al.,2010;Wu et al.,2010;Roeselers et al.,2011)。Roeselers等(2011)报道不同养殖区的鱼类肠道中梭杆菌门类群的丰度差别很大。据此推测,养殖环境中较高丰度的梭杆菌门是其在锦鲤肠道中广泛分布的重要原因。
肠道细菌在协助肠道发挥正常的生理功能方面具有重要意义。鲸杆菌属细菌具有发酵多肽碳水化合物的能力,还可以产生维生素B12(Tsuchiya et al.,2008);气单胞菌和弧菌产生的胞外酶对淀粉和蛋白质的消化起促进作用(Asfie et al.,2000;刘慧玲等,2012);拟杆菌属菌株(包括A型和B型)有水解淀粉和几丁质的能力(Turnbaugh et al.,2006)。与后肠相比,这些菌属在锦鲤前肠和中肠丰度较高,推测与上述2个肠段具有较强的蛋白质和糖类的消化功能有关(Sun et al.,2019)。本研究发现鱼类常见的一些条件性致病菌,如假单胞菌属和黄杆菌属(Wu et al.,2012)具有较低的丰度,其中一个重要原因是肠道微生物中一些正常类群,如鲸杆菌属和拟杆菌属的细菌类群可以抑制病原菌生长繁殖(Sugita et al.,1996,2002),这些菌群在一定程度上保护宿主免受病原菌侵害,起到屏障作用。
据研究报道,由于培养基的营养和生长环境因素的限制,动物肠道微生物中只有一小部分能够在人工培养基上生长(Amann et al.,1995)。本研究中,与16S rDNA高通量测序的鉴定结果相比,应用传统生理生化试验鉴定出的优势菌属较少。另外,一些细菌适合在培养基上生长,其数量往往会被高估(Langendijk-Genevaux et al.,1995),导致培养过程中各类细菌的数量及所占比例发生变化,所以生理生化试验鉴定出的各菌属的丰度与16S rDNA高通量测序的鉴定结果并不一致。即使菌落计数能够得到3个肠段的细菌浓度,但其无法准确地反映肠道不同部位微生物丰度的差异。
4 结论
外部环境可能是影响鱼类肠道菌群的重要因素,这使肠道不同部位中丰度最高的几类菌门相同,也表明养殖水环境中的微生物组成可能对鱼类的生理状态有重要影响。在肠道形态和营养条件等选择压力的作用下,不同部位的菌群结构又表现出不同程度的差异,最终使锦鲤肠道不同部位形成独特的菌群结构,并可能会影响肠道不同部位功能的分化。
参考文献:
李东萍,郭明璋,许文涛. 2015. 16S rRNA测序技术在肠道微生物中的应用研究进展[J]. 生物技术通报,31(2): 71-77. doi:10.13560/j.cnki.biotech.bull.1985.2015.02.010. [Li D P,Guo M Z,Xu W T. 2015. Advances and applications on methodology of 16S rRNA sequencing in gut microbiota analysis[J]. Biotechnology Bulletin,31(2):71-77.]
刘慧玲,罗鹏,杨世平,李广聪,莫嘉文,王蔚. 2012. 具有多種胞外酶的对虾肠道黏附菌的分离和鉴定[J]. 广东海洋大学学报,32(3):1-5. [Liu H L,Luo P,Yang S P,Li G C,Mo J W,Wang W. 2012. Isolation and identification of extracellular enzyme-producing bacteria from the intestinal tract of Litopenaeus vannamei[J]. Journal of Guangdong Ocean University,32(3): 1-5.]
汤伏生,朱晓燕,张兴忠. 1994. 鲤鱼肠道细菌及其淀粉酶对宿主消化的影响[J]. 水产学报,18(3): 177-182. doi:10.1007/BF02007173. [Tang F S,Zhu X Y,Zhang X Z. 1994. The influences of common carp intestinal bacteria and its amylases on the host digestion[J]. Journal of Fishe-ries of China,18(3): 177-1822.]
許文涛,郭星,罗云波,黄昆仑. 2009. 微生物菌群多样性分析方法的研究进展[J]. 食品科学,30(7): 258-265. doi:10.3321/j.issn:1002-6630.2009.07.060. [Xu W T,Guo X,Luo Y B,Huang K L. 2009. Research progress on analysis methods of diversity of microbial flora[J]. Food Science,30(7): 258-265.]
中国科学院微生物研究所细菌分类组. 1978. 一般细菌常用鉴定方法[M]. 北京:科学出版社. [Bacterial Ttaxonomy Group, Institute of Microbiology,Chinese academy of Sciences. 1978. Common identification methods for common bacteria[M]. Beijing: Science Press.]
Amann R I,Ludwig W,Schleifer K H. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J]. Microbiological Reviews,59(1): 143-169. doi:10.1016/S0882-4010(95)90076-4.
Asfie M,Yanagi H,Okano R,Akiyama N,Sugita H. 2000. The protease-producing ability of Vibrios isolated from larvae and juveniles of Japanese flounder[J]. Aquaculyure Science,48(1):139-140. doi:10.11233/aquaculturesci1953. 48.139.
Bowman J P,McCuaig R D. 2003. Biodiversity,community structural shifts,and biogeography of prokaryotes within antarctic continental shelf sediment[J]. Applied and Environmental Microbiology,69(5): 2463-2483. doi:10.1128/AEM.69.5.2463-2483.2003.
Brown K,Decoffe D,Molcan E,Gibson D L. 2012. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease[J]. Nutrients,4(8): 1095-1119. doi:10.3390/nu4081095.
Choi K Y,Lee T K,Sul W J. 2015. Metagenomic analysis of chicken gut microbiota for improving metabolism and health of chickens—A review[J]. Asian Australasian Journal of Animal Sciences,28(9):1217-1225. doi:10.5713/ajas.15.0026.
Ganguly S,Prasad A. 2012. Microflora in fish digestive tract plays significant role in digestion and metabolism[J]. Reviews in Fish Biology and Fisheries,22(1): 11-16. doi:10.1007/s11160-011-9214-x.
Garrity G M. 2005. Bergeys manual of systematic bacteriology[M]. Springer Science & Business Media.
Giatsis C,Sipkema D,Smidt H,Heilig H G H J,Benvenuti G,Verreth J A J,Verdegem M C J. 2015. The impact of rearing environment on the development of gut microbiota in tilapia larvae[J]. Scientific Reports,5(1): 18206. doi:10.1038/srep18206.
Gu S H,Chen D D,Zhang J N,Lü X M,Wang K,Duan L P,Nie Y,Wu X L. 2013. Bacterial community mapping of the mouse gastrointestinal tract[J]. PLoS One,8(10): e74957. doi:10.1371/journal.pone.0074957.
Han S F,Liu Y C,Zhou Z G,He S X,Cao Y N,Shi P J,Yao B,Ring? E. 2010. Analysis of bacterial diversity in the intestine of grass carp(Ctenopharyngodon idellus) based on 16SrDNA gene sequences[J]. Aquaculture Research,42(1): 47-56. doi:10.1111/j.1365-2109.2010.02543.x.
Han Z R,Sun J F,Lü A J,Wang A L. 2018. Biases from different DNA extraction methods in intestine microbiome research based on 16S rDNA sequencing: A case in the koi carp,Cyprinus carpio var. koi[J]. Microbiology Open,8(1): e00626. doi:10.1002/mbo3.626.
Lan C C,Love D R. 2012. Molecular characterisation of bacterial community structure along the intestinal tract of zebrafish (Danio rerio): A pilot study[J]. ISRN Microbio-logy,(4): 590385. doi:10.5402/2012/590385.
Langendijk-Genevaux P,Schut F,Jansen R G C G J,Kamp-huis,G R,Wilkinson M H F,Welling G W. 1995. Quantitative fluorescence in situ hybridization of Bifidobacte-rium spp. with genus-specific 16S rRNA-targeted probes and its application in fecal samples[J]. Applied and Environmental Microbiology,61:3069-3075. doi:10.1128/AEM. 61.8.3069-3075.1995.
Ley R E,Lozupone C A,Hamady M,Knight R,Gordon J I. 2008. Worlds within worlds: Evolution of the vertebrate gut microbiota[J]. Nature Reviews Microbiology,6(10): 776-788. doi:10.1038/nrmicro1978.
Ley R E,Peterson D A,Gordon J I. 2006. Ecological and evolutionary forces shaping microbial diversity in the human in-testine[J]. Cell,124(4): 837-848. doi:10.1016/j.cell.2006. 02.017.
Looft T,Allen H K,Cantarel B L,Levine U Y,Bayles D O,Alt D P,Henrissat B,Stanton T B. 2014. Bacteria,phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations[J]. The ISME Journal,8(8): 1566-1576. doi:10.1038/ismej.2014.12.
Narrowe A B,Albuthi-Lantz M,Smith E P,Bower K J,Roane T M,Vajda A M,Miller C S. 2015. Perturbation and restoration of the fathead minnow gut microbiome after low-level triclosan exposure[J]. Microbiome,3(1): 6. doi:10. 1186/s40168-015-0069-6.
Nayak S K. 2010. Role of gastrointestinal microbiota in fish[J]. Aquaculture Research,41(11): 1553-1573. doi:10. 1111/j.1365-2109.2010.02546.x.
Qin J J,Li R Q,Raes J,Arumugam M,Burgdorf K S,Mani-chanh C,Nielsen T,Pons N,Florence L,Yamada T,Mende D R,Li J H,Xu J M,Li S C,Li D F,Cao J J,Wang B,Liang H Q,Zheng H S,Xie Y L,Tap J,Lepage P,Bertalan M,Batto J M,Hansen T,Le Paslier D,Linneberg A,Nielsen H B,Pelletier E,Renault P,Sicheritz-Ponten T,Turner A K,Zhu H M,Yu C,Li S T,Jian M,Zhou Y D,Li Y R,Zhang X Q,Li S Z,Qin N,Yang H M,Wang J,Brunak S,Doré J,Guarner F,Kristiansen K,Peder-sen O,Parkhill J,Weissenbach J,Bork P,Ehrlich S D,Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature,464 (7285): 59-65. doi:10.1038/nature08821.
Roeselers G,Mittge E K,Stephens W Z,Parichy D M,Cavanaugh C M,Guillemin K,Rawls J F. 2011. Evidence for a core gut microbiota in the zebrafish[J]. The ISME Journal,5(10): 1595-1608. doi:10.1038/ismej.2011.38.
Sugita H,Kawasaki J,Deguchi Y. 1997. Production of amylase by the intestinal microflora in cultured freshwater fish[J]. Letters in Applied Microbiology,24(2): 105-108. doi:10.1046/j.1472-765x.1997.00360.x.
Sugita H,Okano R,Suzuki Y,Iwai D,Mizukami M,Akiyama N,Matsuura S. 2002. Antibacterial abilities of intestinal bacteria from larval and juvenile Japanese flounder against fish pathogens[J]. Fisheries Science,68(5): 1004-1011. doi:10.1046/j.1444-2906.2002.00525.x.
Sugita H,Shibuya K,Shimooka H,Deguchi Y. 1996. Antibacterial abilities of intestinal bacteria in freshwater cultured fish[J]. Aquaculture,145(1-4): 195-203. doi:10.1016/S0044-8486(96)01319-1.
Sun J F,Wang Y Z,Lü A J,Xian J A,Wang Q K,Zhang S L,Guo Y J,Xing K Z. 2019. Histochemical distribution of four types of enzymes and mucous cells in the intestine of koi carp(Cyprinus carpio var. koi)[J]. Fish Physiology and Biochemistry,45(4): 1367-1376. doi:10.1007/s10695- 019-00673-y.
Tsuchiya C,Sakata T,Sucita H. 2008. Novel ecological niche of Cetobacterium somerae,an anaerobic bacterium in the intestinal tracts of freshwater fish[J]. Letters in Applied Microbiology,46(1): 43-48. doi:10.1111/j.1472-765X. 2007.02258.x.
Turnbaugh P J,Ley R E,Mahowald M A,Magrini V,Mardis E R,Gordon J I. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest[J]. Nature,444(7122): 1027-1031. doi:10.1038/nature05414.
Verschuere L,Rombaut G,Sorgeloos P,Verstraete W. 2000. Probiotic bacteria as biological control agents in aquaculture[J]. Microbiology and Molecular Biology Reviews,64(4): 655-671. doi:10.1128/mmbr.64.4.655-671.2000.
Wallace K N,Akhter S,Smith E M,Lorent K,Pack M. 2005. Intestinal growth and differentiation in zebrafish[J]. Me-chanisms of Development,122(2): l57-l73. doi:10.1016/j.mod.2004.10.009.
Wang Y Z,Sun J F,Lü A J,Zhang S L,Sung Y Y,Shi H Y,Hu X C,Chen S J,Xing K Z. 2017. Histochemical distribution of four types of enzymes and mucous cells in the gastrointestinal tract of reared half-smooth tongue sole Cynoglossus semilaevis[J]. Journal of Fish Biology,92(1): 3-16. doi:10.1111/jfb.13469.
Wu S G,Gao T H,Zheng Y Z,Wang W W,Cheng Y Y,Wang G T. 2010. Microbial diversity of intestinal contents and mucus in yellow catfish(Pelteobagrus fulvidraco)[J]. Aquaculture,303(1-4): 1-7. doi:10.1016/j.aquaculture.2009.12.025.
Wu S G,Wang G T,Angert E R,Wang W W,Li W X,Zou H. 2012. Composition,diversity,and origin of the bacterial community in grass carp intestine[J]. PLoS One,7(2): e30440. doi:10.1371/journal.pone.0030440.
(責任编辑 邓慧灵)
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
大众健康(2021年8期)2021-08-04
意林(2021年14期)2021-07-29
科学导报(2021年29期)2021-06-03
意林(2021年3期)2021-03-11
意林(2021年23期)2021-01-16
意林(2019年8期)2019-04-28
科海故事博览·下旬刊(2019年6期)2019-04-16
百科知识(2017年10期)2017-05-19
