
RBP4、α2-MG、SAA在2型糖尿病及其微血管病变中的表达意义
2021-06-30 02:27闫朝丽
重庆医学 2021年11期
王 嫇,闫朝丽
(内蒙古医科大学附属医院内分泌科,呼和浩特 010000)
近年来,我国的糖尿病(DM)患病率逐年上升,以2型糖尿病(T2DM)为主,占所有糖尿病患者的90.0%以上[1]。糖尿病微血管病变是常见的慢性并发症,它包括糖尿病视网膜病变(DR)和糖尿病肾病(DN)。微血管病变的典型改变是微循环障碍和微血管基底膜增厚,目前认为,DN及DR有着共同的发病机制。本课题前期蛋白质组学研究[2-3]发现,新发T2DM组的视黄醇结合蛋白4(RBP4)、α2-巨球蛋白(α2-MG)、血清淀粉样蛋白A(SAA)均表达上调。本研究拟通过测定健康人、合并与未合并微血管病变的T2DM 患者血清RBP4、α2-MG、SAA水平,并收集相关临床指标,对这些因子与T2DM的关系进行验证,并进一步分析其与糖尿病微血管病变的相关性。
1 资料与方法
1.1 一般资料
选择2018年3-9月本院内分泌科收治的T2DM住院患者197例,分为单纯T2DM无微血管并发症组(NMCD组)和糖尿病合并微血管病变组(MCD组)。NMCD组患者132例,男73例,女59例,年龄29~70岁,平均(51.5±8.4)岁;MCD组患者65例,男35例,女30例,其中单纯DR(DR组)28例、单纯DN(DN组)21例、DR合并DN(DR+DN组)16例,年龄35~80岁,平均(57.0±10.0)岁。入选DM及DN患者均符合1999年WHO诊断标准[4],入选DR患者均符合2002年国际临床分级标准[5](排除1型糖尿病、妊娠糖尿病及其他特殊类型糖尿病,并排除糖尿病急性并发症,严重心、脑疾病及外周血管疾病等)。另选取同期本院体检中心健康体检者80例作为健康对照组(NGT组),其中男37例,女43例,年龄31~71岁,平均(51.4±10.0)岁。本研究经本院伦理委员会批准,所有患者均签署知情同意书。
1.2 方法
1.2.1资料收集和实验室检测指标
收集入选患者基本信息、年龄、病程、身高、体重等一般资料,以及实验室检查指标,包括RBP4、空腹血糖(FBG)、空腹C肽、空腹胰岛素(FINS)、糖化血红蛋白(HbA1c)、高密度脂蛋白胆固醇(HDL-C)、低密度脂蛋白胆固醇(LDL-C)、三酰甘油(TG)、总胆固醇(TC)、载脂蛋白A1(ApoA1)、载脂蛋白B100(ApoB100)、纤维蛋白原(Fib)、尿素(Urea)、血肌酐(Cr)、血清胱抑素C(CysC)、尿微量清蛋白/尿肌酐(UACR)、尿微量清蛋白(mAlb)、脂蛋白a[LP(a)]。胰岛素抵抗指数(HOMA2-IR)、胰岛β细胞功能指数(HOMA2-β)用HOMA Calculator v2.2.3版软件据FPG及空腹C肽得出(http://www.dtu.ox.ac.uk/HOMACalculator/)。肾小球滤过率(eGFR)应用CKD-EPI Creatinine 2009 Equation根据血肌酐、年龄、性别、人种计算得出。非HDL-C水平由TC水平减去HDL-C水平得出。
1.2.2人血清α2-MG、SAA测定
取研究对象清晨空腹静脉血3~4 mL至促凝管中,室温血液自然凝固10~20 min,3 000 r/min离心20 min后分离血清,置于—80 ℃冰箱中保存待测。人血清α2-MG、SAA使用南京建成生物工程研究所生产试剂盒,采用酶联免疫法测定。
1.3 统计学处理
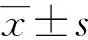
2 结 果
2.1 MCD组中三亚组与NMCD组、NGT组相关指标比较
DR组、DN组、DR+DN组患者性别、年龄、BMI、RBP4、SAA水平比较,差异均无统计学意义(P>0.05),DN组、DR+DN组患者α2-MG水平较DR组高,但均显著高于NGT组,见表1。
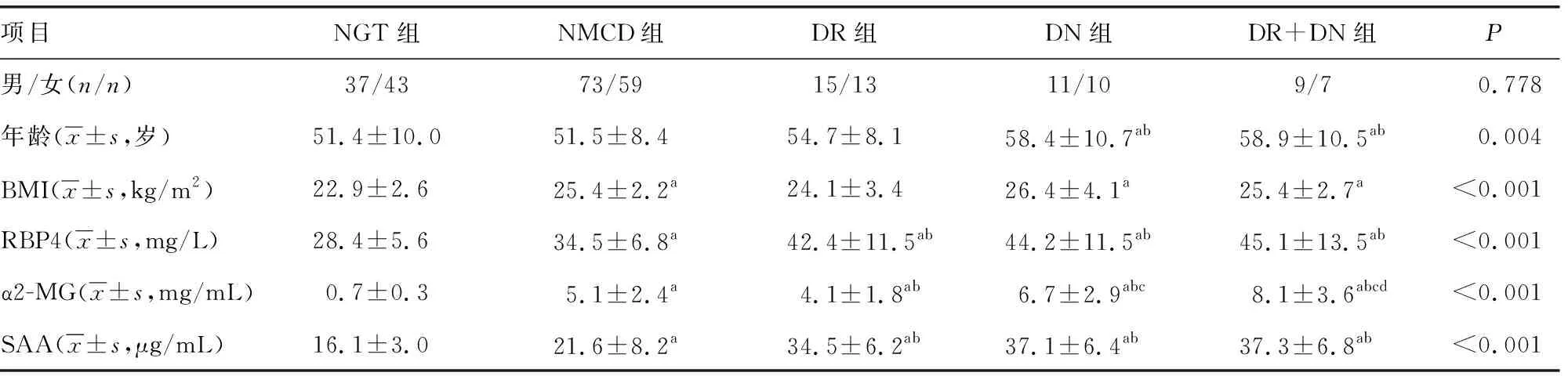
表1 MCD组中三亚组与NMCD组、NGT组相关指标比较
2.2 三组一般资料比较
三组性别构成比差异无统计学意义(P=0.425)。MCD组年龄较另外两组增高(P<0.05),NGT及NMCD组年龄差异无统计学意义(P>0.05)。NMCD组、MCD组BMI、腰围、腰臀比较NGT组高(P<0.05),NMCD组BMI、腰围、腰臀比与MCD组比较,差异均无统计学意义(P>0.05);MCD组病程显著长于NMCD组(P<0.05)。见表2。

表2 三组一般资料比较
2.3 三组RBP4、α2-MG、SAA水平比较
NMCD组、MCD组RBP4、α2-MG、SAA水平较NGT组升高,MCD组RBP4、SAA水平较NMCD组高(P<0.05)。NMCD组α2-MG水平与MCD组比较,差异无统计学意义(P>0.05)。见表3。

表3 三组RBP4、α2-MG、SAA水平比较
2.4 三组间胰岛功能相关指标比较
NMCD组、MCD组FPG及HbA1c较NGT组显著升高(P<0.05),NMCD组与MCD组组间差异无统计学意义(P>0.05);MCD组空腹C肽及HOMA2-β水平较NMCD组显著降低(P<0.05),且NGT组HOMA2-β水平较NMCD组与MCD组显著增高(P<0.05);NMCD组HOMA2-IR较NGT组及MCD组均升高(P<0.05);FINS各组间比较差异无统计学意义(P=0.922)。见表4。
表4 三组间胰岛功能相关指标比较
2.5 三组脂代谢相关指标比较
NMCD组LP(a)、TG水平较NGT组升高(P<0.05),LDL-C、TC、非HDL-C水平与NGT组比较,差异无统计学意义(P>0.05),但有升高趋势。MCD组LDL-C、TC、非HDL-C、ApoB100水平较NMCD组显著升高(P<0.05)。MCD组与NMCD组组间HDL-C、ApoA1、ApoA1/B100比较差异无统计学意义(P>0.05)。见表5。
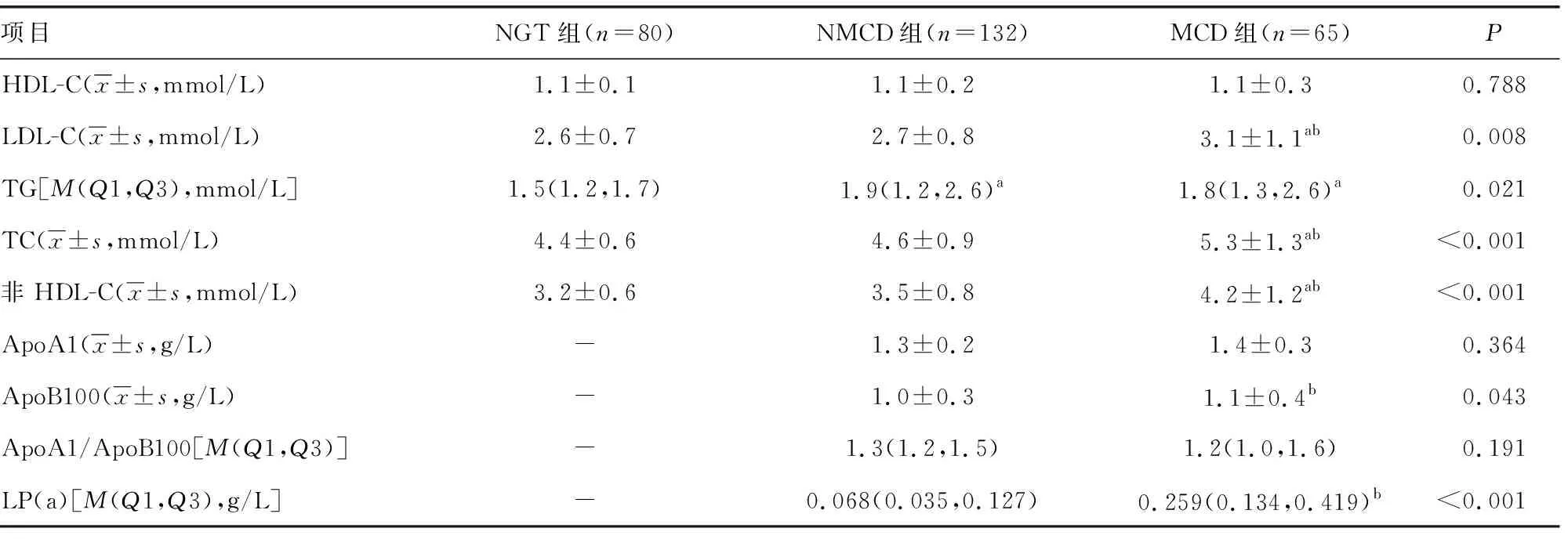
表5 三组间脂代谢相关指标比较
2.6 多因素二分类logistic回归分析
以是否患糖尿病微血管病变为因变量(Y=1或0),以RBP4、SAA、病程、腰围、UACR、Urea、CysC、LDL-C、TC、非HDL-C、ApoB100、LP(a)、mAlb、空腹C肽等为自变量进行二分类logistic回归分析,结果显示,RBP4升高(OR=1.128,95%CI:1.004~1.267,P<0.05)、空腹C肽降低(OR=0.139,95%CI:0.029~0.652),P<0.05)、SAA升高(OR=1.219,95%CI:1.075~1.382,P<0.01)、LP(a)升高(OR=1.073,95%CI:1.017~1.132),P<0.05]是发生糖尿病微血管病变的危险因素。见表6。
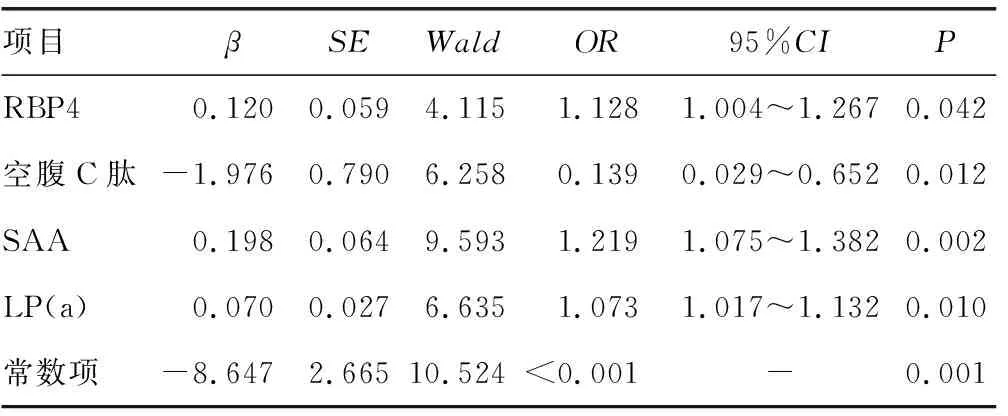
表6 多因素二分类logistic回归分析
3 讨 论
微血管病变是T2DM常见并发症,大量流行病学调查显示,年龄、病程、HbA1c、血脂、HOMA2-IR、蛋白尿等是影响糖尿病微血管病变的重要因素。高血糖产生的糖毒性产物可能通过激活多元醇和己糖通路最终导致血管内皮生长因子(VEGF)的表达增强,从而发生微血管病变。本研究MCD组年龄较另外两组高,随着年龄增长,糖尿病病程延长,发生糖尿病微血管病变风险增加。而多项研究也显示,排除年龄这一因素,RBP4、SAA仍在微血管病变患者中显著增高[6-7]。
RBP4是一种相对分子质量为21 ku的转运蛋白,主要由肝脏合成,也可由脂肪细胞合成与分泌[8]。RBP有几种同种型并在不同的组织中表达,血浆中存在的同种型已知是RBP4[9]。本研究结果显示,RBP4在T2DM患者中显著增高,考虑其与胰岛素抵抗相关的可能机制如下:(1)RBP4 损伤肌肉组织中胰岛素信号传导途径,包括降低磷酸肌醇激酶-3(PI-3K)的活性和抑制胰岛素受体底物-1(IRS1)的磷酸化,从而促使肌肉组织中胰岛素作用受损。(2)RBP4 促进肝糖输出,目前大多认为RBP4直接对肝脏磷酸烯醇式丙酮酸羧激酶(PEPCK)的诱导作用导致其过度表达[10],促进基础状态糖异生,同时又间接通过降低胰岛素抑制肝糖异生的作用对糖代谢造成影响[11],在两方面的共同作用下导致胰岛素抵抗。(3)血清RBP4 通过葡萄糖非氧化途径降低胰岛素敏感性[12]。另外,无论是DN组或DR组,亦或是MCD组,RBP4水平均较NMCD组升高,且二分类logistic回归分析也显示出RBP4与微血管病变的相关性。因为肾脏是主要负责RBP4降解的器官,糖尿病肾病患者系膜细胞增殖、基膜合成加速、肾小球滤过率改变,使RBP4 排泄障碍,可能是RBP4水平升高的原因。同时,本研究也显示,RBP4与一些脂代谢相关指标呈正相关。既往大量研究认为,脂代谢异常等多种成分可直接或者间接引起炎性反应和血管内皮损害,从而引发一系列糖尿病血管并发症,因此也不能除外与RBP4相关的脂代谢异常参与了糖尿病微血管并发症的发生、发展。另外,RBP4不仅转运血液循环中的视黄醇,而且与视网膜上存在的RBP4受体结合后,可将视黄醇释放到视网膜,从而致使大量视黄醇聚集于视网膜上,进而引起功能障碍,导致糖尿病视网膜病变的发生。
SAA 是一种主要由肝细胞合成的急性期蛋白,也是由人脂肪细胞分泌的一种促炎性脂肪细胞因子,其促进人体脂肪组织中的局部炎症并触发全身性慢性低度炎症。本研究结果提示,SAA与胰岛素抵抗存在相关性。目前认为,SAA可通过以下机制导致胰岛素抵抗:T2DM患者多伴有肥胖,脂肪细胞的肥大可导致SAA增加[7],这是因为脂肪组织是巨噬细胞浸润的标靶,巨噬细胞浸润可产生大量炎症因子,SAA是局部和全身的促炎症脂肪细胞因子,它可以刺激中性粒细胞表达和释放大量的炎性因子(TNF-α、IL-6、IL-8等),并发挥细胞毒性作用,引起胰岛β细胞凋亡,导致胰岛素缺乏,胰岛素的不足又反过来促进急性炎性反应的增加,使急性炎性相关产物SAA大量产生,进而形成恶性循环。此外,SAA在胰岛素敏感性下降的个体中呈现高表达,进而激活肝脏、脂肪组织及骨骼肌中的中性粒细胞中的抑制因子、核因子κB、激酶β信号通路,以诱导机体产生胰岛素抵抗。另外有研究发现,SAA可增加脂肪细胞的脂解作用,使脂肪酸的氧化作用增强,进而引起全身的胰岛素抵抗,并对胰岛β细胞产生毒性作用,最终导致胰岛β细胞功能受损,影响糖代谢[13]。同时,SAA可与HDL-C结合促进HDL-C降解,并激活中性粒细胞胆固醇酯酶,使损伤组织产生并释放大量游离胆固醇,引发高胆固醇血症[14]。脂代谢的紊乱可使尿清蛋白排出增加,改变肾小球基膜的磷酸脂成分或引起基膜的葡萄糖胺糖化,使肾小球基膜的通透性增加,使肾小球滤过率下降,这与本研究SAA与UACR、mAlb、CysC呈正相关、与eGFR呈负相关的结果相一致,同时SAA可直接激活丝裂原活化蛋白激酶(MAPK),使肾脏细胞肥大,细胞外基质积聚,导致肾小球硬化和肾小管间质纤维化,这在一定程度上导致和加速了糖尿病肾病的发生。此外,SAA导致的脂代谢紊乱还加重了组织缺血、缺氧,从而诱发和加重微血管的内皮损伤。另外,SAA能参与凝血系统的多个过程,造成凝血系统紊乱,促进血栓形成[15]。以上多种因素的共同作用导致了T2DM微血管病变的发生与发展。
α2-MG是由肝脏产生的一种大分子糖蛋白,本研究结果显示,α2-MG在T2DM患者中显著增高,提示了其与胰岛素抵抗的相关性。有学者认为,这与血清α2-MG可同血清中的胰岛素结合或影响靶细胞对胰岛素的内在化有关,血清中高α2-MG可能会降低胰岛素的生物利用度,导致血糖调节受损,引起胰岛素抵抗[16]。本研究发现,DN组乃至MCD组较NMCD组α2-MG水平显著升高,而DR组较NMCD组α2-MG却无明显增高,考虑原因如下:糖尿病肾病的发病过程多与炎性细胞因子等有关,例如核因子κB会参与糖尿病肾病的发生、发展[17],而核因子κB又是部分α2-MG的激活因子,其通过促进非特异性蛋白酶抑制剂α2-MG的转录合成,从而促使血清α2-MG水平升高。同时,当发生糖尿病肾病时,肾小球基底膜滤过屏障受损,肾小球滤过率增大,血清中升高的α2-MG会漏出体外,因而出现了α2-MG与肾损伤标志相关的结果。可以认为T2DM患者α2-MG水平的增加与糖尿病肾病相关,但不能肯定α2-MG是糖尿病肾病发病的危险因素。此外,有研究证明,α2-MG与氧诱导新血管形成的大鼠视网膜中的低密度脂蛋白受体相关蛋白-1(LRP-1)一起高表达[18],表明这种蛋白酶抑制及其受体LRP-1共同参与视网膜新生血管形成过程,但尚不能认为α2-MG与糖尿病视网膜病变的发生有关。
本研究还发现,MCD组较NMCD组脂代谢紊乱更显著。OUYANG等[19]认为,胰岛素抵抗可能是T2DM合并脂代谢紊乱发生的始动因素,而有研究认为,胰岛素抵抗相关的脂代谢紊乱是导致糖尿病肾病的独立危险因素之一[20]。就单独对于肾脏而言,脂质在肾小球沉积可导致系膜增生,于肾小管中沉积减弱肾小管的重吸收能力,进而产生微量蛋白尿,甚至大分子蛋白从尿液中丢失。另外,微血管病变与血液流变学关系密切,脂代谢紊乱使血液黏稠度增高,导致肾脏微循环障碍,进而引起肾小球血管损伤。同时,脂代谢紊乱会导致胰岛素抵抗加重,增高的胰岛素可导致血管紧张素Ⅱ对肾小球系膜的损伤加强。总之,脂代谢紊乱通过直接或间接作用于肾脏促进了糖尿病肾病的进展。对于糖尿病视网膜病变,脂代谢紊乱引起的血流动力学改变,使眼底血管缺血、缺氧,血管内皮受损,进而出现眼底微血管硬化、渗出、增生、闭塞甚至出血。考虑脂代谢紊乱在微血管病变的发生与发展中起着一定的推动作用。
综上所述,RBP4、SAA升高与胰岛素抵抗及T2DM的发生相关,α2-MG可能与T2DM有关。高RBP4、SAA、LP(a)水平及胰岛功能减退可能是发生T2DM微血管病变的危险因素,而α2-MG在T2DM微血管病变发生、发展中的作用尚不确切。脂代谢紊乱在T2DM微血管病变的发生、发展中起着一定推动作用。但是由于本研究样本较少,仍需继续收集更多的病例,以大样本的研究来进一步证实。
猜你喜欢
中老年保健(2021年5期)2021-08-24
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年3期)2021-07-22
世界最新医学信息文摘(2021年12期)2021-06-09
小雪花·成长指南(2021年2期)2021-05-20
天津医科大学学报(2019年3期)2019-08-13
初中生世界·九年级(2019年4期)2019-05-05
中外医疗(2016年15期)2016-12-01
医学研究杂志(2015年9期)2015-07-01
中国医药导报(2015年26期)2015-02-28
