
基于Gaussian和Multiwfn软件的喹啉和异喹啉的可视化教学研究初探*
2021-06-18 08:16:48杨宝华张爱华
首都师范大学学报(自然科学版) 2021年3期
李 琳,杨宝华,张爱华
(首都医科大学燕京医学院,北京 101300)
0 引 言
喹啉和异喹啉互为同分异构体,是基础有机化学中2个重要的、含有一个杂原子的、六元杂环苯并体系.二者的基本化学性质是吡啶和苯的结合体,苯环上容易发生芳香亲电取代反应,而吡啶环上容易发生芳香亲核取代反应,其分子结构如图1所示.二者分子中苯环与吡啶环上所有π电子形成一个相互重叠的大π体系,但由于氮(N)原子的存在,整个体系的电子重叠不均匀,引起环上不同原子的电子密度分布不均匀,导致不同原子发生芳香亲电取代或亲核取代反应,且发生同种取代反应时存在明显的区域选择性[1-2].因此,在喹啉和异喹啉的教学中,存在化学反应类型和位点多,仅进行文字讲解时,学生对结构和反应位点的区域选择性理解不透彻,容易出现理解混乱,因此,有必要开展直观形象的可视化教学,帮助学生理解二者的性质.

图1 分子结构
Gaussian软件是一个专业的量子化学计算软件,可以完成气相和液相条件下,分子结构优化、分子轨道计算、化学反应和光谱学等诸多方面的计算工作[3].目前许多教师也将Gaussian软件运用到化学领域的相关教学工作中.如周辉和邓萍[4]利用Gaussian软件帮助学生对二甲苯分子的空间结构、振动模式的指认与吸收峰的归属等有更感性的认识,既促进了学生理解红外光谱的本质,又提高了学生的学习兴趣和积极性;侯彦君等[5]利用Gaussian软件研究了分子红外光谱计算、振动模式指认以及同位素取代在分子振动光谱中的应用,帮助学生深入思考同位素取代对红外特征吸收峰的影响;刘佳等[6]利用Gaussian软件模拟了几种溶剂对溶质分子的结构与红外光谱造成的影响,深化学生对溶剂效应本质的认识,加深了对红外光谱中溶剂效应的理解;王光琴等[7]阐述了Gaussian软件在太赫兹(tera hertz,THz)光谱模拟方面的研究情况;苟高章等[8]利用Gaussian软件帮助学生了解手性分子的三维空间结构、电子圆二色谱的理解及归属等;赵一丹和廖奕[9]借助 Gaussian软件对氢气(H2)分子进行了势能面扫描,描绘了其能量曲线,比较了价键法和分子轨道法计算得到的H2电子云分布,将复杂的线性变分法解波函数简单化.可见,Gaussian软件在教学中的有效应用,丰富了教学手段,有利于教学重难点的突破,提高教学质量.
Multiwfn软件是波函数分析程序,通过波函数分析方法对化学问题进行分析,利用交互式的操作,获取对研究化学问题有益的信息[10].韩晓刚和廖奕[11]将Multiwfn和Gaussian软件结合应用于结构化学教学,通过分析三重态H2分子拉伸过程的定域化轨道指示函数的填色图和电子密度差等值线图,说明了H2分子的电子结构特点.
与Multiwfn软件相比,Gaussian软件在实际教学中的应用较多,而目前尚无基于二者的有关喹啉和异喹啉的可视化教学研究的论文.为了直观形象地解释二者性质的异同,并用于课堂教学,解决实际教学中存在的难点,本文尝试利用不同方法探讨喹啉和异喹啉的性质差异[12].利用密度泛函理论(density functional theory,DFT)、等化学屏蔽表面(iso⁃chemical shielding surfaces,ICSS)、电子定域化函数(electron localization function,ELF)和 Mulliken布居分析方法对二者的性质进行可视化分析.
1 ICSS法
1996 年,Schleyer等[13]提出核独立化学位移(nucleus⁃independent chemical shifts,NICS)理论,是目前应用的比较广泛的表征具有各种环状离域电子的化合物芳香性的磁性依据.采用量子化学的方法计算分子的绝对屏蔽值,用于分析化合物是否具有芳香性.NICS计算的是特定点上或一条线上磁屏蔽值的变化,或研究特定平面上磁屏蔽值的分布,常用指标包括计算环几何中心位置的NICS(0)值、计算环平面上下0.1 nm位置的NICS(1)值和垂直于环平面(ZZ)方向的 NICS_ZZ值等[13-14].当外磁场导致的体系共轭环上的环电流产生的感应磁场对外磁场有屏蔽作用时,会削弱外磁场强度,屏蔽值是正值,NICS取屏蔽值的相反数,为负值,体系为芳香性体系;当体系环电流的感应磁场和外磁场方向相同,对外磁场起去屏蔽作用,会加强外磁场强度,屏蔽值为负值,NICS为正值,体系为反芳香性体系.NICS值越负,表明该位置对外磁场屏蔽程度越大,该环具有的芳香性越强;NICS值越正,说明对外磁场的去屏蔽程度越大,反芳香性越强;NICS值接近于0,则认为该体系无芳香性.
ICSS是在三维层面展现空间不同位置的磁屏蔽值[15],故在符号取值上与NICS值符号相反.收集三维空间不同位置的磁屏蔽张量信息绘制成ICSS图像,使得ICSS与无法可视化的NICS方法相比,以图像化的形式,直观展现体系的化学性质.
本文绘制喹啉和异喹啉的ICSS的等值面图和等值线图所用的方法,与文献[16]中描述蒽和菲ICSS值所用的方法一致.
1.1 ICSS等值面
在ICSS等值面图上,ICSS值变化时,可以得到屏蔽和去屏蔽区域不同的ICSS等值面,随着ICSS值增大,去屏蔽的等值面会明显收缩直至断开,然后屏蔽值小的区域会出现孔洞,从而展现出分子中不同位置的屏蔽能力.
喹啉的ICSS等值面如图2所示.当ICSS=11.0 ppm和 ICSS_ZZ=28.0 ppm(1.0 ppm=1.0×10-6)时,苯环的等值面被覆盖得较明显,吡啶环的等值面中心开始出现凹陷;当ICSS=11.5 ppm时,吡啶环上的孔洞明显比苯环上的孔洞大;当ICSS_ZZ=29.0 ppm时,吡啶环上明显存在孔洞,而苯环还是被屏蔽值较大的等值面覆盖.说明苯环和吡啶环的等值面明显不同,苯环对外磁场的屏蔽能力更强,芳香性比吡啶环更显著.
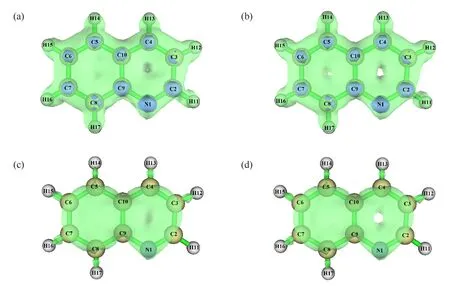
图2 喹啉的等化学屏蔽表面(ICSS)等值面
异喹啉的ICSS等值面如图3所示.当ICSS=11.0 ppm时,吡啶环稍有凹陷;当ICSS=11.5 ppm时,苯环和吡啶环均出现明显的孔洞,但吡啶环上的孔洞明显更大;当ICSS_ZZ=28.0 ppm时,二者的等值面均被覆盖的较明显;当ICSS_ZZ=29.0 ppm时,在吡啶环上出现了明显的孔洞,而苯环上还是被屏蔽值较大的等值面覆盖着.可知,苯环和吡啶环的等值面明显不同,苯环对外磁场的屏蔽能力更强,与喹啉一致,苯环的芳香性也比吡啶环更显著.

图3 异喹啉的ICSS等值面
分析ICSS等值面,在喹啉和异喹啉分子中,苯环上的ICSS屏蔽值比吡啶环上大,苯环的电子云密度略高一些.因此,苯环的芳香性比吡啶环强.
1.2 ICSS_ZZ等值线
ICSS_ZZ屏蔽值的大小与色彩刻度的深度呈正相关,即红色越深的区域,ICSS屏蔽值越大.喹啉和异喹啉的ICSS_ZZ等值线如图4所示.在喹啉的环平面上方0.1 nm处,所有C原子附近红色深浅不同,说明不同C原子附近ICSS值不同,也间接表明π电子分布并不均匀.如果不考虑苯环和吡啶环共用的碳碳键(C—C),除了吡啶环上N1的屏蔽值较大以外,其他C原子的屏蔽值几乎都比苯环上C原子的屏蔽值小,尤其与N1相邻的C2原子的屏蔽值明显更小.这说明吡啶环上N原子的电子密度较大,而C原子,尤其与N原子邻近的C原子的电子密度较低,吡啶环电子密度集中在N原子附近,电子离域程度不如苯环,使得吡啶环的芳香性降低.这与喹啉的苯环易于发生硝化、磺化和卤代反应等亲电取代反应,而吡啶环上的C原子,尤其是与N原子相邻的C原子更易发生亲核取代反应的性质一致;而N原子屏蔽值大,电子密度大,易于结合质子,显碱性[1-2].
与喹啉类似,在异喹啉环平面上方0.1 nm处,不同C原子ICSS值亦不同,π电子分布不均匀.吡啶环在N2原子处出现了较高的屏蔽值,而C原子的屏蔽值比苯环上C原子的屏蔽值小,吡啶环上电子的离域程度稍差,芳香性比苯环的芳香性弱.因此,异喹啉的吡啶环上也易于发生亲核取代反应,苯环上易于发生亲电取代反应;而N原子屏蔽值大,电子密度大,也易于结合质子,显碱性.
从以上分析可知,ICSS等值面和等值线图的优势各不相同.ICSS等值面图在分析喹啉和异喹啉分子不同环的屏蔽值及芳香性差异时,直观方便;而ICSS等值线图在分析不同原子上的屏蔽值更具优势,且其可间接地分析电子密度是否分布均匀,从而探讨不同原子的反应活性.
2 ELF法
ELF 是 Becke和 Edgecombe[17]与 Jordi等[18]提出的表征电子定域性特征的实空间函数,是量子化学领域中研究电子结构特征的重要工具,其能展现三维实空间中不同位置的电子定域程度.ELF方法在三维空间对电子云密度选取等值面的值不同时,得到不同的等值面范围.ELF值的范围为0~1.ELF等值面的值越大,其等值面内的空间区域就越小,随着ELF值的增大,各个原子的离域空间最终都会被断开,形成每个原子相对独立的电子云区域.ELF值与物质的芳香性关系为:在ELF值较低时,出现独立的区域,等值面包围的区域小,电子分散程度低,在该区域离域范围小,芳香性弱;在ELF值较高时,出现独立的区域,等值面包围的区域大,电子在等值面内分散程度大,在该区域离域性好,区域稳定性高,即电子能够稳定地在该区域内离域,芳香性强[12,19-21].
喹啉和异喹啉的芳香性质主要是由π电子多中心离域引起,因此,计算时仅考虑对芳香性质有贡献价值的π电子,得到ELF⁃π值图.二者的ELF⁃π二分点数值图的操作步骤与文献[16]一致.
2.1 ELF⁃π等值面
增加ELF⁃π等值面数值,会逐渐展现出喹啉和异喹啉体系不同环上键断开能力差异(即能体现出芳香性差异).不同ELF⁃π值时喹啉和异喹啉的等值面如图5所示.当ELF⁃π=0.70时,喹啉的吡啶环上C9—N1键发生断裂,基本不参与共轭体系,而苯环没有出现断键;当 ELF⁃π=0.75时,其等值面在C9—C10、C9—C8、C10—C5、C10—C4和 C9—N1键处均断开,整个喹啉分子断裂成4个域,包括苯环和吡啶环上的2个π电子4中心离域结构和C9、C10上2个单原子域.由以上分析可知,在ELF⁃π值较低时,喹啉的吡啶环首先发生断裂,说明喹啉中苯环比吡啶环的等值面包围的区域大,电子离域程度大,且芳香性更强.

图5 不同ELF⁃π时等值面
当ELF⁃π=0.70时,异喹啉的苯环和吡啶环上的等值面还是各自连通的,但C3—N2键连接紧密程度降低,说明吡啶六元环在等值面数值更低时会首先断开;当 ELF⁃π=0.75时,C9—C8、C9—C10、C10—C5、C10—C4、C9—C1及C3—N2键均断开,异喹啉的苯环上包含了1个显著的4中心离域结构,而吡啶环上孤立的域更多,这些均体现了苯环上的π电子离域程度明显高于吡啶环,因此,芳香性比吡啶环强,这一点与喹啉是一致的.
2.2 ELF⁃π二分点数值
为了更精确地得到原子形成相对独立的电子云区域时,化学键刚好断开的ELF值,利用ELF⁃π拓扑分析得到喹啉和异喹啉的ELF⁃π二分点数值如图6所示 .喹啉分子中,C9—C10、C9—C8、C10—C5、C10—C4、C9—N1和 C2—N1的ELF⁃π二分点数值分别约为 0.73、0.74、0.74、0.75、0.65和 0.93,其中吡啶环上C9—N1发生断裂的二分点值最小,即最易发生键断裂,其比苯环上C—C断裂数值均低.与苯环相比,吡啶环的电子离域程度更低,且芳香性更弱 .异喹啉分子中,C9—C10、C9—C8、C10—C5、C9—C1、C10—C4、C3—N2和 C1—N2的二分点数值分别约为 0.73、0.74、0.74、0.75、0.73、0.71和 0.94,其中吡啶环中C3—N2发生断裂的二分点数值最小.与喹啉类似,吡啶环的芳香性更弱.

图6 ELF⁃π拓扑分析的二分点数值
3 Mulliken布居分析
原子电荷是描述化学体系中电荷分布最简单、最直观的方式之一.Mulliken布居是Mulliken提出的计算电荷在分子中各组成原子之间分布情况的方法,分析分子的原子电荷及电荷在各原子轨道上的分布等情况[22].芳香性物质的亲电取代反应易发生在电子云密度大的原子上,而亲核取代反应易发生在电子云密度小的原子上,因此,喹啉和异喹啉分子中,电荷越负(或越正)的原子越有可能吸引亲电(或亲核)试剂进攻,而发生亲电(或亲核)取代反应[23-25].由于喹啉和异喹啉都是含有苯环和吡啶环的体系,π电子及相应的π轨道与其反应活性关系最为密切.因此,可排除σ电子的影响,仅利用π轨道Mulliken电子布居数和π电子密度预测喹啉和异喹啉的性质[19].
启动Multiwfn软件,绘制出利用Mulliken布居分析计算的等值面值为0.035时,喹啉和异喹啉的π电子密度等值面,详细如图7所示.喹啉和异喹啉上N原子的π电子密度明显较大,但不同C原子上的π电子密度差异不大.计算不同原子的Mulliken电子布居数,喹啉分子中,N1、C2、C3、C4、C5、C6、C7和C8原子的Mulliken电子布居数分别为1.088、0.962、0.996、0.990、1.030、0.988、0.989和 0.972;异喹啉分子中,C1、N2、C3、C4、C5、C6、C7和C8原子的Mulliken电子布居数分别为 0.975、1.093、0.993、1.012、1.019、0.975、0.995和1.001.
图7 Mulliken布居分析的π电子密度等值面
因为不同C原子的π电子数差异不大,所以电子密度等值面图不能清晰地展示出其电子密度的差异.因此,绘制了分子平面上方0.1 nm处的π电子密度填色图(图8),可用于直观地分析喹啉和异喹啉发生芳香亲电和亲核取代反应的可能位点.由π电子密度的色彩刻度可知,喹啉分子中,与其他C原子相比,C2和C4原子的橘色范围小、颜色浅,说明这2个位置的电子密度明显比其他位置的电子密度低,即C2和C4位置易发生芳香亲核取代反应;与C4原子相比,C2原子的橘色范围更小、颜色更浅,说明C2比C4原子的电子密度更低,即C2位置更易发生芳香亲核取代反应.这与计算的Mulliken电子布居数是一致的,C2原子的Mulliken电子布居数为0.962,是喹啉分子中最小的.这一结果与文献中喹啉的芳香亲核取代反应易发生在吡啶环C2和C4原子上,且C2原子的取代产物多于C4原子的取代产物的结论一致[1].对应C5原子的Mulliken电子布居数为1.030,是C原子中Mulliken电子布居数最大的,说明C5原子的电子密度最大,可知喹啉的亲电取代反应最易发生在苯环上,并且容易生成C5原子上的取代产物,也与文献的结论一致[1].

图8 Mulliken布居分析的π电子密度填色
异喹啉分子中,与其他C原子相比,C1原子的橘色范围最小,颜色最浅,Mulliken电子布居数为0.975,是吡啶环原子中电子密度最小的,是最易发生亲核取代反应的位置.这与文献中描述异喹啉的亲核取代反应主要在C1原子上发生、几乎没有C3位置产物的结论一致[1].对应C5原子的Mulliken电子布居数为1.019,是所有C原子中电子密度最大的,是亲电取代反应最易发生的位点.
对于喹啉和异喹啉分子中的N原子而言,其橘色范围均是最大且颜色最深.二者N原子的Mulliken电子布居数分别为1.088和1.093,均为最大值,说明其电子密度均最大,易接受质子或与Lewis酸配位,显示碱性.而与喹啉分子中的N原子相比,异喹啉分子中N原子的Mulliken电子布居数略大,说明其电子密度略大,更易吸引质子,这与异喹啉的碱性比喹啉的碱性略强也一致[1-2].
4 结束语
本文将Gaussain和Multiwfn软件结合,采用IC⁃SS、ELF和Mulliken布居分析等方法对喹啉和异喹啉的化学性质进行了可视化分析,且相关结果直观地解释了二者的化学性质.结果表明,Gaussain和Multiwfn软件结合可清楚地展示喹啉和异喹啉中苯环的芳香性均强于吡啶环,亲电取代反应易于发生在苯环上,亲核取代反应易发生在吡啶环上,并很好地解释了喹啉和异喹啉的化学反应位点,与文献的结论一致[1-2].这一可视化分析结果使基础有机化学中有关芳香杂环化合物喹啉和异喹啉的教学过程实现可视化成为可能,为学生掌握和理解其芳香性和反应位点提供了辅助依据,同时为具有芳香性的杂环化合物的教学提供思路和借鉴,使学生对基础有机化学芳香杂环化合物的化学反应原理和基本理论产生更加深入的认识,有效地提高教学质量.可视化教学是对教材内容有益的深化和补充,在教学实践中合理地利用化学软件辅助教学,可以为理解化学问题提供更多的方法和手段,有助于学生直观形象地理解化学问题,理解物质的本质,激发学生的学习兴趣.
猜你喜欢
大学化学(2021年8期)2021-09-26 10:50:46
国防科技大学学报(2020年6期)2020-12-07 09:25:48
山东化工(2020年5期)2020-04-07 09:59:30
测绘通报(2019年11期)2019-12-03 01:47:34
赤峰学院学报·自然科学版(2019年5期)2019-09-10 07:22:44
心肺血管病杂志(2019年4期)2019-06-27 07:36:10
测绘学报(2018年1期)2018-02-27 02:23:07
数学理论与应用(2016年4期)2016-05-17 04:50:26
小学生导刊(低年级)(2016年2期)2016-02-24 22:39:09
应用数学与计算数学学报(2014年4期)2014-09-26 12:15:53
