
蒽加氢产物的结构指认和定量核磁共振分析
2021-06-09 00:52:36窦梦雨侯相林唐明兴王英雄
波谱学杂志 2021年2期
窦梦雨,赵 奇,侯相林,刘 雷,唐明兴,王英雄
1. 中国科学院 山西煤炭化学研究所,山西 太原 030001;2. 中国科学院大学,材料与光电研究中心,北京 100049;3. 太原理工大学 化学化工学院,山西 太原 030024
引 言
稠环类芳烃化合物(如蒽、菲、萘)广泛存在于煤油、石油、汽油等各类油品中.这些稠环类芳烃化合物不仅降低了油品的十六烷值,导致油品的重度劣质化,而且其不完全燃烧会排放出 CO等有毒气体和苯等致癌物质.通过对稠环芳烃催化加氢处理,从而降低油品中稠环芳烃的含量以获得清洁燃料油,可显著降低油品使用过程中对环境的污染,使其符合目前国家对油品燃料标准的要求[1-5].
蒽(AN)作为稠环芳烃的代表物之一,其催化加氢产物包括二氢蒽(DHA)、四氢蒽(THA)、对称八氢蒽(sym-OHA)、非对称八氢蒽[(cis+trans)OHA]和全氢蒽(PHA)(图1)[6].通过调变催化剂及控制催化条件选择性的获得高附加值产物,是当前蒽催化加氢的研究热点之一[7-11].例如,该反应中获得的对称八氢蒽进一步生成均苯四甲酸二酐,它是重要的化工中间体,可用于环氧树脂固化剂、聚酰亚胺原料、聚酯单体及热塑性增塑剂等领域[12,13].由于蒽催化加氢产物沸点接近,从中分离出对称八氢蒽十分困难且能耗巨大,所以通过调整催化条件提高产物中对称八氢蒽的含量是更为经济合理的途径.但是蒽催化加氢反应历程长、反应本身复杂,且产物结构相似、极性相近,导致该反应的产物分析十分困难.

图1 蒽加氢反应路径Fig. 1 Reaction pathways for the anthracene hydrogenation
目前,文献[1,3,6,8]报道的蒽催化加氢产物分析多采用气相色谱法和气质联用法.色谱法分析混合物的局限性在于其不能给出详细的分子结构信息,且不能解析未知产物结构.此外,由于蒽催化加氢产物沸点普遍较高、分子量相差小、结构相近,在使用气质联用仪进行定性分析时产物荷质比与质谱数据库对比相似性不高.所以寻求一种准确、快速、有效的分析方法,鉴定产物结构进而指导优化蒽加氢反应条件、提高产物的选择性是十分有必要的.
核磁共振(NMR)是一种非侵入性和非破坏性的检测技术,由于可实现多维、多核检测,因此NMR谱图通常具有较高的分辨率,在复杂混合物的分析方面展现出诸如方便快捷、分辨率高、无需标准物质比对等优势,这是其它常规分析检测技术所不具备的[14,15].近年来,二维扩散排序谱(DOSY)、一维选择性激发谱(selTOCSY)、纯化学位移谱(pure shift)等NMR高级方法已被逐渐用于混合物的分析[16-19].我们团队通过使用DOSY和selTOCSY方法,成功分析了甘油加氢反应和生物质平台化合物加氢/氢解反应的产品混合物;使用pure shift技术成功检测出了费托合成反应生成水中的11种含氧化合物[20-23].Ma等[21]在混合物的分析中首先使用DOSY技术对扩散系数有明显差异的组分(如:正丙醇、正丁醇和甲醇)进行了结构定性,再使用selTOCSY技术通过激发1H NMR谱图中确定的信号峰进一步确认了扩散系数相近的组分(如乙酸和乙醇).Lyu等[23]联合使用DOSY和selTOCSY技术鉴定了5-羟甲基糠醛、2,5-二(羟甲基)呋喃、2,5-二(羟甲基)四氢呋喃等多种化合物.这些前期的研究成果表明,DOSY联合selTOCSY技术在结构相似、分子量相近的混合物体系的结构分析中表现出强大的优势.本文先通过 DOSY、selTOCSY结合一系列常规NMR方法(1H NMR、13C NMR、DEPT135、1H-1H COSY、1H-13C HSQC)对蒽加氢反应中不同加氢程度的三个样品进行了分析,然后使用定量核磁共振氢谱(QNMR)计算得到了蒽的转化率和产物的选择性,为稠环芳烃及其催化加氢产物的分析提供了新的选择.
1 实验部分
1.1 试剂与反应
氘代氯仿(CDCl3,99.8 atom% D)由剑桥同位素实验室提供;吡嗪(C4H4N2,98%分析纯)和蒽(C14H10,质量分数≥99.5%)均购于上海阿拉丁试剂生化科技股份有限公司;环己烷(C6H12,分析纯)购于天津市科密欧化学试剂有限公司;四氢呋喃(C4H8O,分析纯)购于国药集团化学试剂有限公司;四甲基硅烷(TMS)购于东京化成工业株式会社.
蒽催化加氢反应均使用Pd负载量为1%(w/w)的Pd/Al2O3作为催化剂.反应1的反应溶剂为环己烷,50 ℃下反应3 h;反应2的反应溶剂为四氢呋喃,200 ℃下反应3 h;反应3的反应溶剂为环己烷,先在50 ℃下反应3 h,然后升温至100 ℃再反应3 h.三个反应均在3.0 MPa的氢气压力下、50 mL的反应釜中进行,反应结束后快速降至室温,离心并烘干后,分别得到固体样品1、2、3(包括产物及反应物蒽),取固体样品进行NMR测试.
1.2 样品准备
除QNMR实验外,其余NMR实验中各取2.0 mg固体样品溶于400 μL CDCl3,转移至直径为5 mm的NMR样品管待测,以TMS定标(δH0.00,δC0.0).
QNMR样品制备:分别准确称取一定量的样品1、样品2、样品3置于三个不同的离心管中,向三个离心管中各加入一定量的内标物吡嗪和400 μL CDCl3,振荡溶解,转移至NMR样品管中待测.
1.3 NMR实验
1D和2D NMR实验均在Bruker Avance 400 MHz NMR波谱仪(配备5 mm PABBO BB/19F-1H/D变温探头)上完成.
1H、13C NMR的工作频率分别为400.13和100.62 MHz,实验温度为25℃.谱宽分别为8 012.820和24 038.461 Hz.selTOCSY实验的混合时间为80~120 ms.
二维谱包括2D梯度场1H-1H COSY、1H-13C HSQC和DOSY.3个样品1H-1H COSY的两维谱宽[F2(1H)和F1(1H)维谱宽相同]分别为3 225.806、3 937.008和3 205.128 Hz,采样数据点阵均为t2×t1=2 048×128,累加次数均为4,测试时间为19.3 min;HSQC的F2(1H)和F1(13C)维的谱宽分别为4 795.396和16 668.479 Hz,采样数据点阵均为t2×t1=2 048×256,累加次数均为4,测试时间为39.6 min;DOSY扩散时间(Δ)为160 ms,梯度恢复延迟时间为0.2 ms,涡流延迟时间为5 ms,梯度强度从 2%到 60%,测试时间为 23.7 min.通过 Bruker Topspin3.1、MestReNova 9.0.1以及Dynamics Center 2.2.4等软件处理数据和谱图.
为了保证QNMR谱图的准确性,设置循环等待时间D1≥5T1.因为待分析物(固体样品包含的化合物)与吡嗪不发生反应,且待分析物的1H NMR谱峰与吡嗪谱峰不重叠,所以我们选择吡嗪作为QNMR实验的内标.采用(1)式计算蒽加氢催化反应产物中各组分的含量[24,25]:

其中I(x)和I(std)分别为样品中待测组分和内标物(吡嗪)定量峰的面积,nH(x)和nH(std)分别为待测组分和内标物定量峰包含的质子数,MW(x)和MW(std)分别为待测组分和内标物的相对分子质量(吡嗪:80.04;蒽:178.08;二氢蒽:180.09;四氢蒽:182.11;八氢蒽:186.14;单位均为g/mol),P(x)和P(std)分别为待测组分和内标物的质量分数(吡嗪:98%),w(x)和w(std)分别为待测组分和内标物的称样量(单位为g).QNMR数据处理使用了GSD(Global Spectral Deconvolution)算法.每份样品平行测定5次,取平均值.
2 结果与讨论
2.1 1H NMR谱图分析
对于当前研究的蒽催化加氢反应,当改变催化反应条件后,所得产物的组成及其含量是不同的.表S1(可扫描文章首页OSID码或在文章网页版查看)中列出了蒽催化加氢反应可能出现的六种化合物的结构式.三个样品的1H NMR谱图在芳香区(aromatic area)[图2(a)]和脂肪区(aliphatic area)[图2(b)]均有信号.二氢蒽在脂肪区有一个信号(-CH2),在芳香区有两个化学位移不同的信号(-CH);四氢蒽在脂肪区有两个化学位移不同的信号(-CH2),而在芳香区有3个化学位移不同的信号(-CH);对称八氢蒽在脂肪区有两个化学位移不同的信号(-CH2),而在芳香区仅有一个单一信号(-CH);非对称八氢蒽和全氢蒽由于具有多种立体异构,因此其脂肪区信号峰比表S1中平面型结构式所表现出的质子种类(非对称八氢蒽4种,全氢蒽4种)多,且更加复杂[26];原料蒽仅在芳香区有三组信号.由于各成分的化学结构相近,在1H NMR谱图的芳香区出现了多组峰型相同、化学位移接近的信号,在脂肪区出现了信号峰重叠的现象.通过1H NMR谱图很难指认各产物,并对各产物质子进行准确归属,从而无法准确鉴别催化加氢产物.因此,需要结合DOSY和selTOCSY技术对样品进行精确分析.
2.2 样品1的组成及其含量分析
DOSY技术根据混合物扩散系数的差异可以在扩散维度上对混合物中的各组分进行虚分离[27,28],我们尝试进行了样品1的DOSY实验(图3).反应产物、反应溶剂环己烷(cyclohexane)、氘代试剂(CDCl3)和 TMS在扩散维上得到了很好的分离.所有的反应产物表现出两个扩散系数D1=1.29×10-9m2/s,D2=1.47×10-9m2/s.D1在1H NMR 谱上对应三个信号:δH6.78、δH2.70 和δH1.77,其中δH6.78为苯环区次甲基(-CH)的信号峰,δH2.70和δH1.77可能为苯环加氢后的亚甲基(-CH2)信号峰,此外,根据COSY谱(图S1)给出的信息:δH2.70和δH1.77信号相关,表明其在结构式中为相邻碳上的氢原子.根据分子结构信息(表S1),对称八氢蒽分子中心对称,满足含有处于三种不同环境的质子(一个芳香区次甲基,两个苯环加氢后的亚甲基)的要求,据此我们将D1对应的三个信号确定为对称八氢蒽.D2对应的1H NMR信号多且复杂,显然包含两种以上产物,从DOSY谱图上未能完全分开.

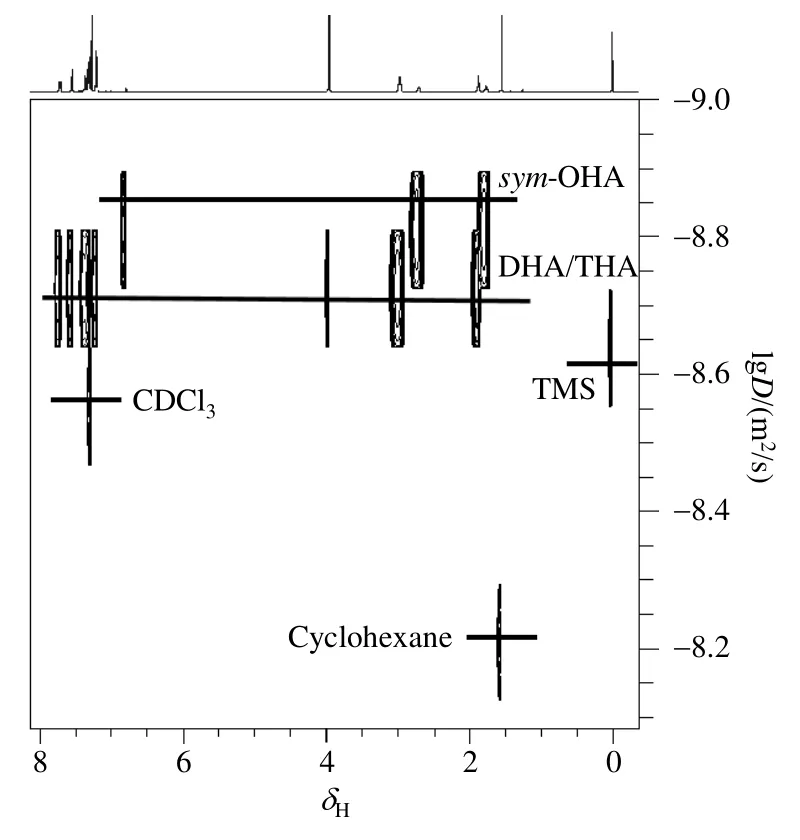
图3 样品1的DOSY谱图Fig. 3 DOSY spectrum of sample 1
为进一步分析产物组成,我们测试了样品1的selTOCSY谱图.selTOCSY技术通过激发选定的质子信号峰,将与激发核耦合的峰从复杂的NMR谱中提取出来,从而得到目标化合物谱图或部分谱图[29].在DOSY技术对混合样品中的信号进行初步指认并提供参考信息后,通过selTOCSY方法激发已确定的信号峰,可以进一步辨别重叠谱峰.我们通过选择性激发δH7.53和δH3.93的两处信号峰,获得了两套完整的亚谱(sub-spectrum)(图4).激发δH7.53的信号得到的亚谱有五个信号峰(δH7.34、δH7.70、δH7.53、δH2.97、δH1.86),其中δH7.34、δH7.70 和δH7.53 推测为苯环区的次甲基(-CH)信号,δH2.97和δH1.86为苯环加氢后的亚甲基(-CH2)信号.在COSY谱图(图S1)中,δH7.34和δH7.70相关,δH2.97和δH1.86相关.结合分子结构信息(表S1)以及谱峰裂分情况,可以确定激发δH7.53获得的亚谱为四氢蒽的完整1H NMR谱图.激发δH3.93的信号从混合谱中提取出了两个与其质子耦合的苯环区次甲基(-CH)信号峰(δH7.29和δH7.19),且这两个信号峰在COSY谱中相关(图S1),满足二氢蒽的分子结构信息(表S1),所以我们将激发δH3.93获得的亚谱确定为二氢蒽的完整1H NMR谱图.
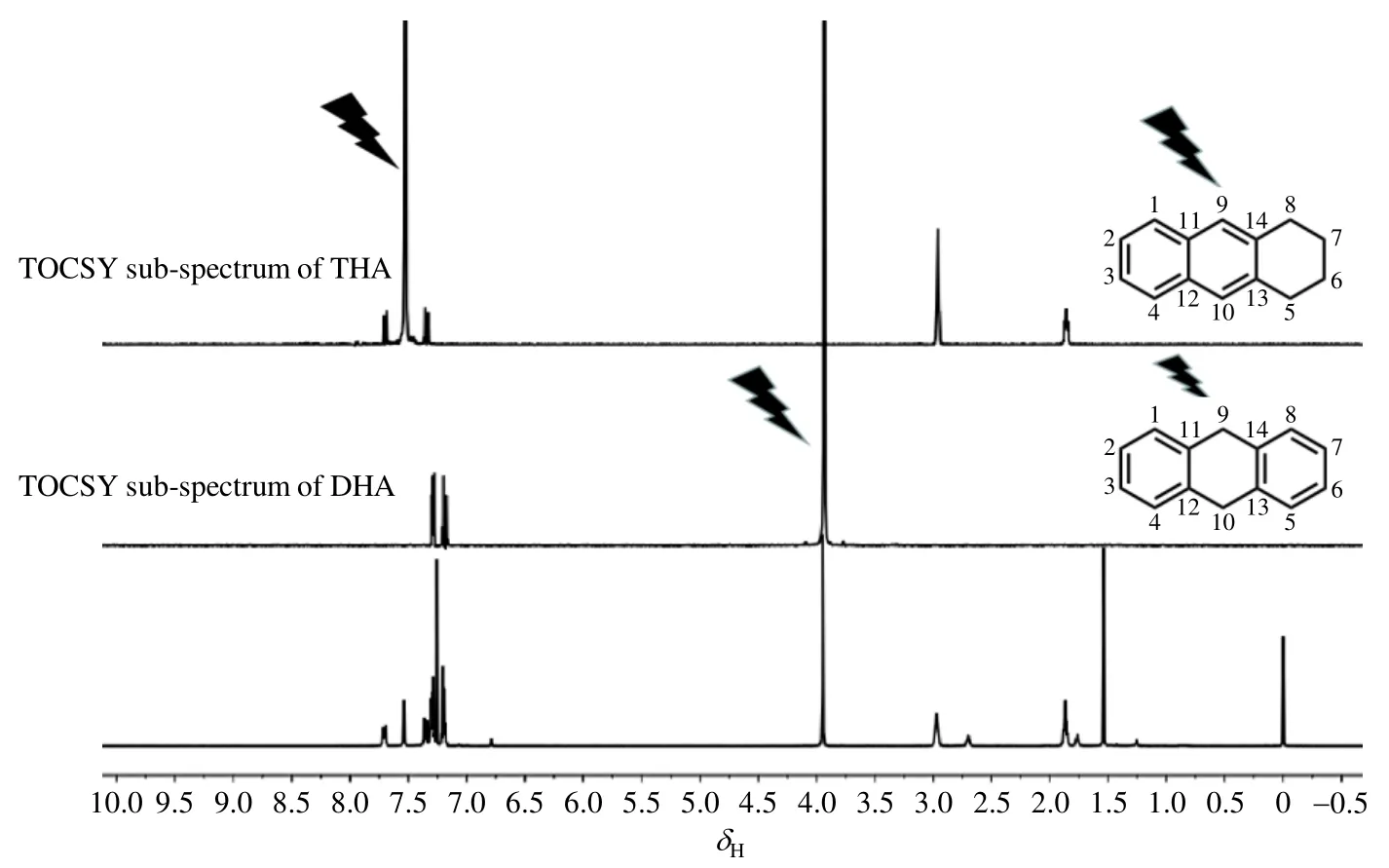
图4 样品1的selTOCSY谱图Fig. 4 selTOCSY spectra of sample 1
联合DOSY和selTOCSY技术初步确定了样品1中含有二氢蒽、四氢蒽和对称八氢蒽.在此信息指导下,我们通过1H-1H COSY(图S1)、DEPT135(图S2)、13C NMR(图S2)和1H-13C HSQC(图S3)实验,对产物中各化合物进行了详细的谱峰归属(表1).
在以上分析的基础上,我们通过QNMR[图S4(a)]对样品1中各组分进行了定量计算(表2),得到了蒽的转化率(100%)和各产物的选择性(二氢蒽:50.73%;四氢蒽:42.80%;对称八氢蒽:6.47%).
我们根据NMR分析结果反馈指导催化条件优化,以提高对称八氢蒽的选择性.根据上述分析结果,我们认为在样品1的催化反应条件下,虽然蒽的转化率达100%,但对称八氢蒽的含量并不高,其选择性仅为6.47%.因为蒽在四氢呋喃中溶解度较高,为了提高对称八氢蒽的选择性,我们尝试在催化剂不变的情况下将反应溶剂由环己烷更换为四氢呋喃,并在200℃的温度下制备了样品2.

表1 蒽加氢产物的1H NMR和13C NMR数据归属Table 1 1H NMR and 13C NMR data assignments of anthracene hydrogenation products
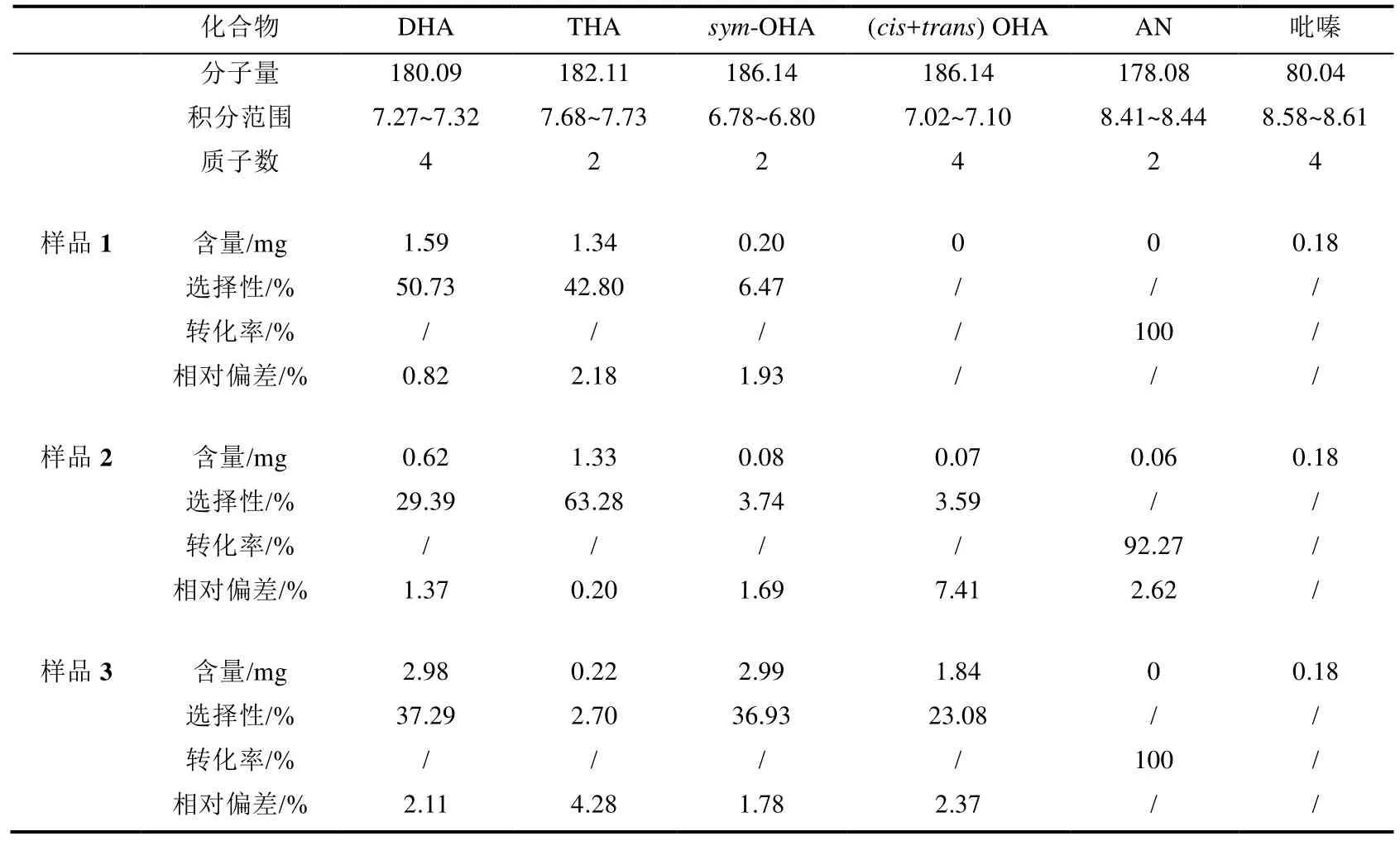
表2 各样品的定量核磁共振氢谱测试分析Table 2 Quantitative analysis for QNMR tests of the 3 samples
2.3 样品2的组成及其含量分析
通过对比样品2和样品1的1H NMR谱(图2),确定了样品2中含有二氢蒽、四氢蒽和对称八氢蒽.根据文献[30],我们在样品2中发现了少量的蒽(δH8.42、δH8.00和δH7.45,表1),说明在样品2的催化反应条件下,原料蒽没有反应完全.
此外,样品2的1H NMR谱图(图S5)的高场区有五组明显的强谱峰信号,根据1H-1H COSY(图S6),我们对其进行以下归属:δH3.64(4H,td,J=6.3/3.4 Hz)和δH1.66(4H,m)归属为反应溶剂四氢呋喃,δH4.32(2H,t,J=7.0 Hz)、δH2.47(2H,dd,J=8.7/7.8 Hz)和δH2.23(2H,m)归属为γ-丁内酯.我们推测在催化反应后的溶剂后处理过程中四氢呋喃可能部分转化为γ-丁内酯[31].
QNMR[图S4(b)]计算结果(表2)表明,样品2中蒽的转化率为92.27%,二氢蒽、四氢蒽、对称八氢蒽的选择性分别为29.39%、63.28%、3.74%.与样品1相比,样品2蒽的转化率和对称八氢蒽的选择性均下降.溶剂由环己烷换成四氢呋喃并提高反应温度后,蒽的转化率和对称八氢蒽的选择性均降低,说明四氢呋喃抑制了蒽的加氢及对称八氢蒽的生成.并且四氢呋喃在后续脱溶剂过程中极易氧化生成聚合物[30],导致产物体系的分析和分离都更加困难.所以我们认为环己烷更适合作为蒽加氢反应的溶剂.于是在样品1的基础上升高反应温度至100 ℃再反应3 h,制得样品3.
2.4 样品3的组成及其含量分析
通过样品3和样品1的1H NMR谱峰对比(图2),可以确定样品3中含有二氢蒽、四氢蒽和对称八氢蒽.另外,由于产物中非对称八氢蒽和全氢蒽具有多种立体异构,因此在样品3的1H NMR谱图中(图S7),高场区(δH1.35~2.83)有多个耦合峰,在δH7.06处也出现了强耦合峰,COSY谱图(图S8)也无法提供有效的相关信息.于是我们使用selTOCSY技术激发了δH7.06处的信号峰,得到了一套完整的亚谱(图5).δH7.06是苯环区次甲基(-CH)的信号峰,全氢蒽是蒽完全加氢后的产物,不再含有苯环,所以激发δH7.06提取出来的子谱可能为非对称八氢蒽的氢谱.

图5 样品3的selTOCSY谱图Fig. 5 selTOCSY spectra of sample 3
样品3的QNMR[图S4(c)]计算结果(表2)表明,蒽的转化率为100%,二氢蒽、四氢蒽、非对称八氢蒽的选择性分别为 37.29%、2.70%、23.08%.最重要的是比较样品1和样品3的QNMR结果后发现:在保持蒽完全转化的情况下,对称八氢蒽的选择性由样品1中的6.47%提高到了样品3中的36.93%.
对比样品2和样品3的1H NMR谱图(图2),发现样品2的1H NMR中δH7.06处也出现了非对称八氢蒽在低场区的特征峰,然后我们又激发了样品2在δH7.06处的信号峰,得到了selTOCSY谱图(图S9),从而我们推测样品2中也含有少量的非对称八氢蒽(选择性3.59%).
3 结论
本文利用DOSY联合selTOCSY技术确定了二氢蒽、四氢蒽、对称八氢蒽和非对称八氢蒽等蒽催化加氢的产物.然后使用一系列常规NMR方法,包括1H NMR、13C NMR、DEPT135、1H-1H COSY、1H-13C HSQC对二氢蒽、四氢蒽和对称八氢蒽进行了详细的1H和13C NMR谱峰归属;并通过QNMR计算了产物的选择性和蒽的转化率.利用NMR分析结果指导并调整了催化反应条件,提高了产物中高附加值组分对称八氢蒽的选择性.以上分析证实NMR技术可以准确、快速的对蒽催化加氢反应产物体系进行定性和定量分析,并且所得的结果可以指导改变催化反应条件以调整反应主要产物的分布.本研究工作对蒽催化加氢反应的研究具有重要意义,同时为稠环类芳烃催化加氢产物的分析提供了系统的NMR技术方案.
利益冲突
无
附件材料
表S1 蒽催化加氢反应的产物体系中可能涉及到的化合物.
图S1 样品1的(a)高场和(b)低场区域放大的1H-1H COSY谱图.
图S2 样品1的DEPT135和13C NMR谱图.
图S3 样品1的(a)高场和(b)低场区域放大的1H-13C HSQC谱图.
图S4 (a)样品1、(b)样品2和(c)样品3的QNMR谱图.
图S5 样品2的1H NMR谱图.
图S6 样品2的1H-1H COSY谱图.
图S7 样品3的1H NMR谱图.
图S8 样品3的1H-1H COSY谱图.
图S9 (a)样品2的1H NMR谱图;(b)样品3的selTOCSY谱图;(c)样品2的selTOCSY谱图.
猜你喜欢
化工设计(2022年4期)2023-01-02 17:44:05
数学年刊A辑(中文版)(2018年2期)2019-01-08 01:59:50
化工管理(2017年35期)2018-01-10 11:19:56
数学理论与应用(2016年4期)2016-05-17 04:50:23
电测与仪表(2015年4期)2015-04-12 00:43:04
化学工业与工程(2015年1期)2015-02-10 03:01:37
中国酿造(2015年4期)2015-01-26 22:50:40
火炸药学报(2014年1期)2014-03-20 13:17:22
中成药(2014年10期)2014-02-28 22:29:32
无机化学学报(2014年9期)2014-02-28 17:33:07
