
二氧化碳电还原反应的理论研究
2021-06-02 11:39:26苑琦杨昊谢淼程涛
物理化学学报 2021年5期
苑琦,杨昊,谢淼,程涛
苏州大学功能纳米与软物质研究院,江苏 苏州 215123
1 引言
二氧化碳电还原反应(CO2RR)是目前全世界都关注的重要研究课题1-4,其重要性主要体现在三个方面。
第一,通过电化学反应还原二氧化碳(CO2)对于保护环境具有积极作用。温室效应是大气保温效应的俗称。二氧化碳作为一种典型的温室气体,自工业革命以来,人类向大气中排入的二氧化碳等吸热性强的温室气体逐年增加(如图1所示),大气的温室效应也随之增强。温室效应会导致一系列连锁的气候问题。如愈加频繁出现的极端气候,如洪水、高温等,很可能都是温室效应的直接后果。尤其需要强调的是,从化学的角度来看化学反应的速率对温度的依赖性大致上遵循指数关系,而指数关系的特点是,初期增长不明显,但是一旦超过临界值将会出现不可控的局面。所以,我们要时刻认识到,温度升高对人类的影响并非简单的线性关系。所以,早日控制住二氧化碳在空气中的含量对人类正常生活是至关重要的。
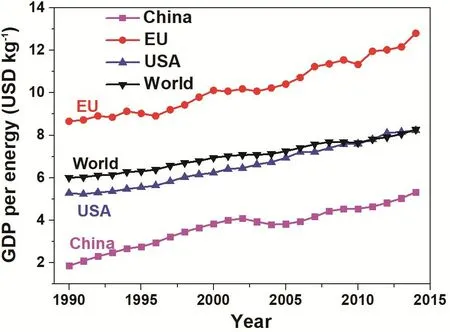
图1 世界国内生产总值(按购买力以2011年美元计算)相对于能源消耗与二氧化碳排放量增长的关系8Fig. 1 World GDP (purchasing power parity, 2011 dollars) is growing relative to energy consumption and CO2 emissions.
第二,通过电化学反应还原二氧化碳或可实现电能的化学储存。随着可持续能源技术的发展,将可再生能源转化为电能的技术已经日趋成熟5-7。随之而来的问题就是如何将电能储存起来。通过电化学反应利用电能将大宗廉价的化学品转化为高附加值的产品,也就实现了电能的化学储存。通过这种方式可以把二氧化碳变废为宝。降低二氧化碳含量的同时,也增加了其商业价值。
第三,二氧化碳电还原反应也是一种很好的模型反应,具有重要的科学研究意义。与其他被广泛研究的电化学反应,如氢气还原、氧气还原等反应不同(二氧化碳电还原,涉及到多电子转移,并且反应会产生多种还原产物),对其反应机理的研究有利于理解其他复杂电化学反应。
二氧化碳电还原的问题在于,其产物分布广泛且选择性差。从热力学角度来看,多种二氧化碳还原产物在热力学上都是可行的。假定二氧化碳为气体状态、水为液体状态、并且产物也按照其室温下的稳定状态为参考态。那么,二氧化碳还原产物对应的平衡还原电势可以根据气相的热力学数据结合依据亨利定律推算出的液体数据进行理论预测。根据美国国家标准技术研究所(NIST)的数据,我们对多种还原产物的平衡电位进行了预测,其结果在表1中。
从表1中不难看出,大部分二氧化碳电还原产物在热力学上是可行的。这也解释了实验中观测到的二氧化碳电还原的多种产物的现象9。那么在这些产物中,哪些产物是更有研究价值的呢?这可以从两方面考虑:一方面是产物本身具有的经济价值,另一方面是产物的合成路线的复杂性10-12。
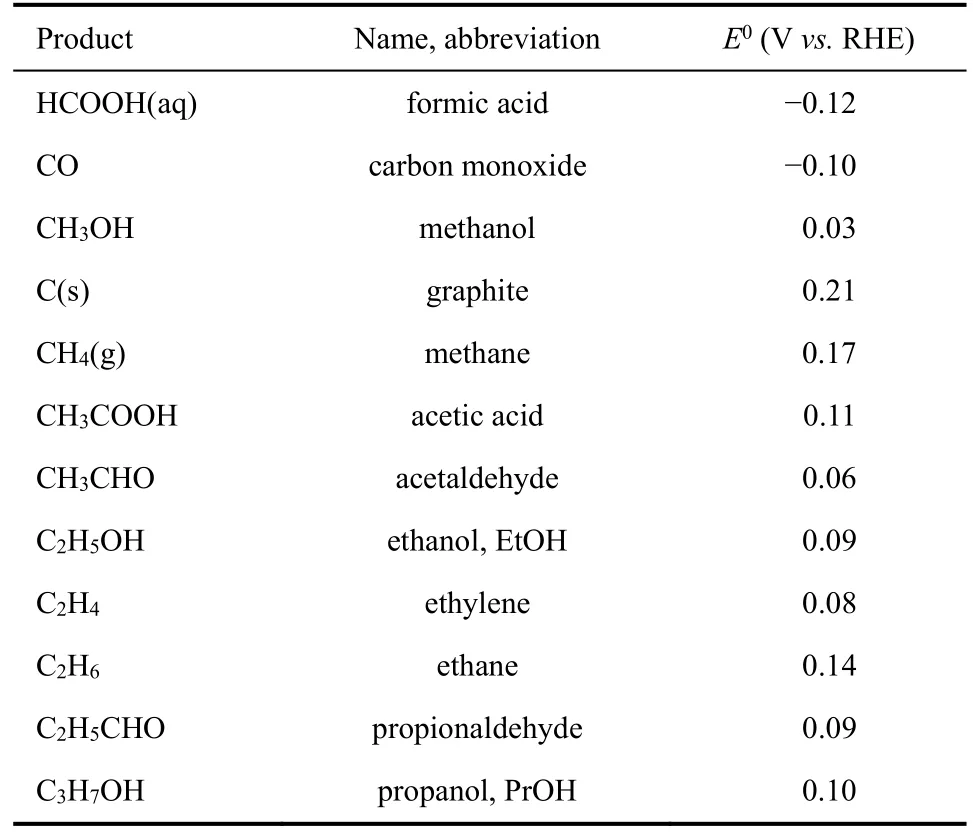
表1 电化学反应的平衡电位Table 1 Electrochemical reactions with equilibrium potentials.
从经济价值来看,乙烯和乙醇是比较理想的产物。显然,产品的市场价格越高,经济价值也越高。如果仅以价格来看,乙酸和丙醇的市场价格较高,似乎是比较理想的产物。但是,值得注意的是,工业需求量也是一种重要的考量标准。虽然乙酸和丙醇的市场价格高,但是其工业需求较少。如图2所示,综合上述两种考虑,乙醇和乙烯一方面价格较高,另一方面工业需求量大。因此乙醇和乙烯是理想的产物。
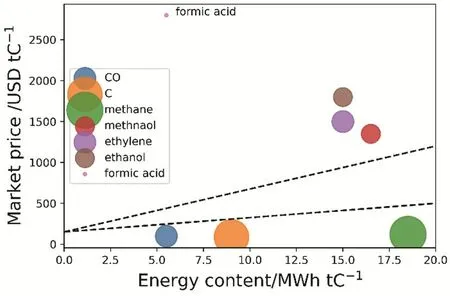
图2 二氧化碳还原产物的市场价格与能量含量的关系8Fig. 2 Market price of select CO2 recycling products as a function of energy content.
如果考虑分离的成本,乙烯产物相较于乙醇产物更为理想。室温下,乙烯处于气体状态,且溶解度低。因此,非常容易从电解液中分离出来。而乙醇的分离则需要额外的能源输入,并相应的需要增加分离步骤。最后,从能量产物角度来看,乙烯也是一种理想的产物。所以,美国能源部提出以乙烯为主要的目标产物。因此,我们认为乙烯是一种最优的产物是建立在综合多方因素考虑的结果。
事实上,针对任何一种产物的专一选择性也是具有研究意义和工业价值的。例如,纯态一氧化碳(CO)也具有比较高的市场价格,并且其合成相对乙烯也较为容易,已有文献报道其高选择性。此外,丙醇是最接近燃油的二氧化碳还原产物,具有非常高的燃烧值和燃烧稳定性,几乎可以直接用于替代汽车燃油,并且其溶解度也较低易于分离。
二氧化碳电还原的核心问题是催化剂的选择13。虽然多种二氧化碳电还原的产物在热力学上是可行的。但是,二氧化碳是比较稳定的分子,激活其碳―氧双键需要耗费大量的能量。因此,通过催化剂活化反应中间体,降低反应能垒是解决二氧化碳电还原的关键。
虽然很多金属都可以实现二氧化碳电还原,但是只有铜(Cu)可以催化还原二氧化碳得到深度产物。早在上个世纪八十年代,日本科学家Hori等人就发现,很多金属都可以实现二氧化碳还原14,15。根据还原产物可以将金属分为四类(如图3所示):第一类是可以将二氧化碳还原为甲酸(或甲酸盐)的金属,包括铅(Pb),汞(Hg),铊(Tl),铟(In),锡(Sn),镉(Cd)和铋(Bi);第二类是可以将二氧化碳还原为一氧化碳的金属,包括金(Au),银(Ag),锌(Zn),钯(Pd)和镓(Ga);第三类是低效催化剂,仅能少量转化二氧化碳(产生一氧化碳),大部分电能转变为氢还原,这类金属包括:镍(Ni),铁(Fe),铂(Pt)和钛(Ti)。最后一类,也就是第四类,仅有一种金属,就是铜(Cu),除了两电子产物CO和甲酸,铜(Cu)还可以把二氧化碳,通过多电子还原(即深度还原)得到碳氢类产物,如甲烷和乙烯等17-19。所以,从还原效能角度来看,对于二氧化碳还原,铜是一种非常特殊的金属(如图4所示)。
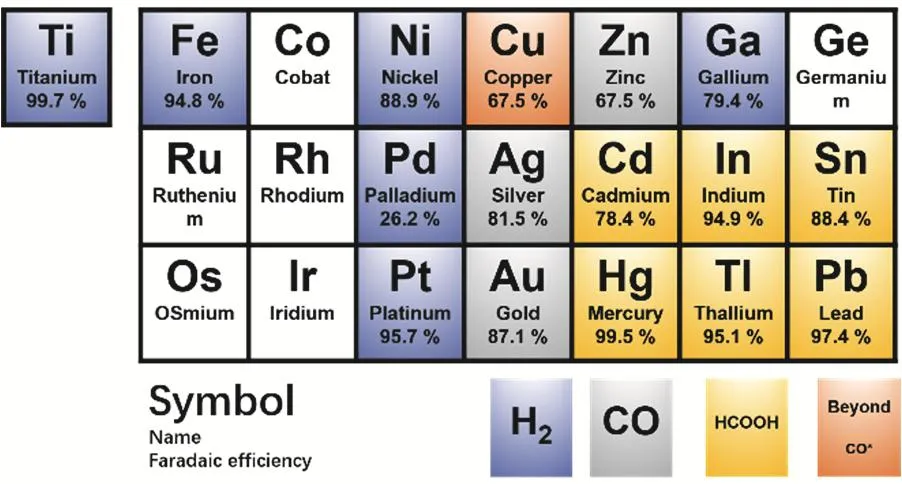
图3 二氧化碳还原金属的分类16Fig. 3 CO2 reduction metal classification.
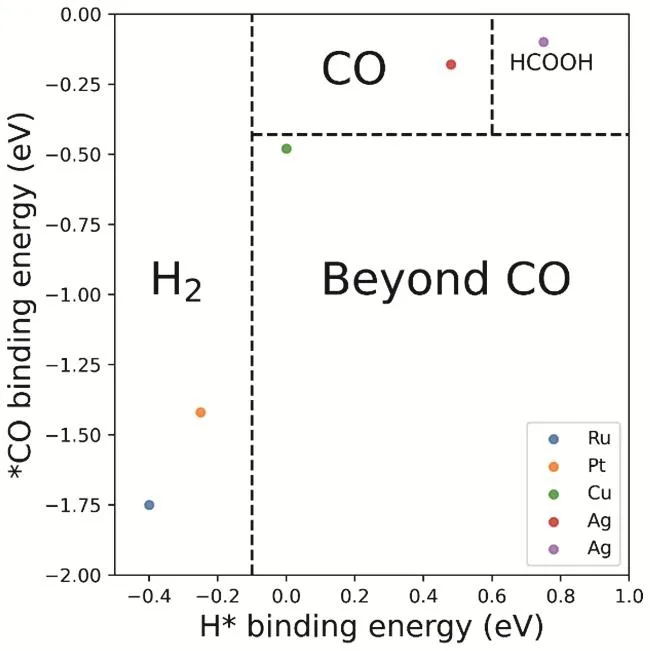
图4 二氧化碳还原产物与吸附能的相关性16Fig. 4 The relationship between CO2 reduction metal classification and binding energies.
尽管可以获得深度产物,铜的催化性能远远无法满足工业应用的要求。一方面,二氧化碳还原在铜体系中的过电位过高,提高了制备成本。另一方面,在二氧化碳还原电位下,氢还原反应(HER)也非常迅速,这导致了大量的H2副产物的生成,造成了能量的浪费。最后,铜的催化选择性差,主产物的生成伴随着大量的副产物,给后续的产物分离造成了困难。所以,加速催化剂的开发,实现高效和高选择性催化剂是目前人们努力攻克的难题。
高效催化剂的理性开发依赖于明确反应机理的研究。理论模拟计算在研究反应机理方面具有明显的优势20。首先,基于原子水平的量子力学方法可以从原子和分子水平对反应的微观机理进行研究。目前实验方法还很难提供原子和分子水平的解析度。因此,理论计算方法为反应机理的研究提供了原子水平的理论依据。其次,理论计算中的模拟模型完全可控,可以实现针对指定材料、指定晶面的研究,这也是实验方法很难做到的。最后,理论计算可以实现高速、大范围的材料扫描。从而实现材料的快速筛选,为实验合成提供可靠的参考意见。综上所述,理论模拟方法在电化学反应机理方面具有明显的优势,是实验的有效补充。理论计算结合实验是解决催化机理研究和材料设计的有力手段21。
在本文中,我们将以铜(Cu)为重点,总结近些年来在反应机理研究方面的研究成果。首先,我们将对常用的理论模拟方法进行总结。然后,我们将对铜催化二氧化碳的重要产物的反应机理进行总结。接下来,我们将结合原位实验观测结果与理论预测进行比较,筛选出与实验数据相符的理论预测机理。在此基础上,我们将探讨提高催化剂性能的合成策略,包括纳米催化剂的活性位点和金属表面改性等方法。最后,我们将介绍新的二氧化碳合成研究思路和机器学习等方法在催化剂机理研究和开发方面的应用。
2 理论计算方法
理论预测结果的准确性依赖于理论方法的准确性。总的来说,理论方法的准确性包括两方面:一方面,理论方法要能够准确的描述原子间相互作用和化学反应的能量。另一方面,理论模拟中的模拟模型要与实验体系保持一致。以下将分别对上述两方面进行讨论。
2.1 密度泛函理论
量子化学方法是目前在研究非均相催化中应用最为广泛的研究手段。量子化学方法可以准确的描述原子间相互作用和化学反应能量。在不依赖于实验数据的情况下,也能提供可靠的预测结果。在多种量子化学方法中,基于密度泛函理论(DFT)的计算方法,是一种最为常用的方法。这是因为密度泛函能够在可以承担的计算量下,给出可靠的预测结果。其可靠性,已经得到了大量的证实。在二氧化碳电化学还原的研究中,密度泛函方法也是应用最为广泛的方法。
在密度泛函计算中,交换关联泛函的选择非常关键22。在众多方法中,我们推荐采用基于广义梯度近似的PBE方法。
此外,密度泛函方法在处理弱相互作用方面存在不足。相应的,我们推荐在计算中包含vdW校正项。密度泛函方法中vdW的校正有两种方式,第一种是在交叉关联泛函中考虑。第二种是采用经验势函数的方法进行后期校正。第一种方法虽然理论上严格,但是往往计算量过于昂贵。所以,目前应用并不广泛。第二种方法,由于计算量低,并且其参数是利用准确的实验结果校正而得,其准确性也有保障。因此,第二种方法是比较流行的vdW校正方法。在众多方法中,比较有代表性的方法是D3和lg。
当然,上述方法并不是最完美的解决方案。在一些情况下,现有理论方法存在着明显不足。例如,实验结果表明,CO在Cu表面的吸附为on-top吸附。但是PBE的预测结果是CO在Cu表面fcc位点吸附。上述差异反映了PBE的不足23。此外,有大量的证据表明D3 vdW 校正高估了金属和吸附物种的相互作用。尽管现有理论框架存在不足,但是在大多数情况下表现良好。并且,由于误差消除等内在因素,最终的预测结果与实验结果相当。因此,我们认为,上述选择是在现有计算能力下的最优解决方法。
2.2 溶剂化效应
电化学反应总是在溶液中进行。溶剂化效应对于反应中间体的构象、稳定性有重要影响。因此,在计算中需要考虑。早期的工作中,人们采用真空计算+后校正的处理方法。上述方法虽然能够一定程度上考虑溶剂化效应,但是其准确性严重不足。并且,由于实际计算是在真空条件下进行,有时会出现构象预测错误的情况。这极大的影响了计算的准确性。
目前比较常用的溶剂化处理方法是隐式溶剂化模型24-26。隐式溶剂化模型把溶剂环境简单地当成可极化的连续介质进行考虑,优点在于其计算量相对较小。
采用显式溶剂化方法考虑溶剂化效应是目前最完备的解决方案。需要注意的是,采用显式溶剂化方法的计算量比较昂贵。
2.3 电压效应
电化学反应的特点是需要施加电压。在二氧化碳电化学还原中需要施加一个负电压驱动反应的进行。那么在理论模拟中,如何模拟电压效应呢?目前常用的处理方式有以下几种:
第一,CHE模型。CHE模型是由Norskov等人提出的电化学计算模型27。该模型的贡献在于通过引入H电极有效的将化学方面转化为电化学反应。由于其易用性,该模型被广泛的应用于电化学反应的研究中。该方法的缺陷在于:第一,仅能反应物和产物进行校正,对于反应过程中的电荷转移无法考虑。第二,该方法属于后校正方法,无法估计界面物种对于施加电压的响应。
第二,巨正则系综模型。电化学反应对应于电子开放体系,也就是说反应中伴随着电荷数目的改变。在一般的量子化学模拟中,电荷数量是固定的,这与电化学反应不符。CHE模型通过后处理的方式解决了反应物和产物的电荷平衡问题。但是无法考虑反应的中间状态。为了全面的考虑电化学反应,需要在模拟过程中根据电化学势,动态的调整电荷数量来保证恒定的外部电压,也就通常所说的巨正则系综模型。Taylor等人发现了外加电荷与能量的二次关系,并将其广泛的应用于电化学反应的研究中28。但是,对于周期性体系来说。外加电荷会导致能量不收敛的问题29。因此,该方法在初期并没有得到广泛的应用。直到近些年来,人们在显示溶剂的开发中,利用壳电荷模型解决了点电荷收敛问题30,31。由于其理论的完备性,目前这一方法越来越多地被应用于电化学反应模拟中。
值得注意的是,目前也有很多后处理校正的方法32,33。需要特别指出的是,虽然推导过程略有区别,但是这些方法在本质上与巨正则系综模型是一致的。在应用这些模型时,建议利用电容(具有明确物理意义的参量)取代电荷(无明确物理意义)来消除由于电荷估计造成的巨大误差。
第三,杂化模型。外加电势的改变显然会对电极界面的分子取向造成影响。相应的,在模拟中利用显式溶剂分子模拟电化学界面是十分必要的。在显式溶剂中,可以通过调控阳离子的数目来改变外加电压。该方法的好处在于:由于溶液的屏蔽效应,解决了真空中的净电荷能量不收敛的问题。但是该方法也存在明显的不足:第一,受限于阳离子只能整数地引入,因此无法实现电势的连续调控。第二,由于阳离子引入过多对造成阳离子浓度过高,太过于偏离实验条件。
解决上述问题的方法是采用显式-隐式杂化模型。杂化模型结合了两者优势:利用显式模型来描述电化学界面,利用隐式模型连续调控电势。因此,能够在接近实验条件的情况下进行模拟。大幅度提升了理论预测的准确性。虽然,目前该方法依然存在计算量昂贵的局限性。但是,可以预见的是,由于其准确性和高自动化水平,将成为未来计算的主流方法。
2.4 pH效应
pH对于电化学反应具有明显的影响34,35。以二氧化碳电还原反应来说,实验pH需要控制在7左右。这是考虑了CO2溶解平衡的结果。大量的实验证据表明,只有CO2可以被还原,KCO3和HCO3则不能36。pH也对选择性造成了影响。在CO还原中,实验研究发现在低pH的情况下,HER反应会大量的发生。而CO还原则相对较弱。在高pH情况下,HER被抑制,CO还原增强。并且产物中C2占优。pH直接影响了绝对电压。在实验中,为了考虑pH的影响,需要将标准氢电极(SHE)电压校正为可逆氢电极(RHE)电压。其转换关系为0.592。值得注意的是,体系中部分物种对于绝对电压敏感。所以,改变pH会导致选择性的变化。相应地,在模拟中考虑pH的影响是十分必要的。
在模拟中,pH的效应从两方面体现。一方面是溶剂中的H3O+和OH-浓度。该效应只能通过显式溶剂模拟。但是,受限于量子化学昂贵的计算量。目前,仅能考虑高浓度情况,例如pH = 0和pH= 14。另一方面是pH带来的电压效应。该效应可以在多种模型中考虑。
2.5 分子动力学模拟
实际体系中,电化学界面处于动态。分子动力学模拟可以考虑界面动态变化的过程,以及温度、同位素效应等动力学因素。通常来说,静态结构优化和过渡态搜寻是非均相催化中被广泛应用的研究方法。但是,越来越多的证据表面,催化剂在反应条件下的动态变化是理解催化机理和催化性能的关键因素。因此,分子动力学模拟越来越被广泛的应用于化学反应的研究中。
2.6 自由能计算
理论上,如果分子动力学模拟的时间足够长,可以实现化学反应的直接模拟。但是,受限于现有计算量的约束,目前基于量子化学的分子动力学模拟所能达到的时间尺度在1 ns以内。而实际的化学反应时间尺度在秒(s)左右。所以,目前的模拟水平和实际反应的时间要求存在9个数量级的鸿沟。以现有的理论计算水平,无法实现对电化学反应的直接模拟。为了弥补上述模拟中的困难,需要采用一些高级的取样方法。
目前比较常用的高级取样方法包括,Metadynamics方法37、限制分子动力学方法38-41和加速分子动力学方法等。这些方法通过引入有偏势,可以定向的驱动反应的发生。从而实现在有限的时间尺度模拟化学反应的目的。高级取样方法结合杂化模型可以实现在接近实验条件下对电化学反应进行理论模拟。
2.7 动力学蒙特卡洛方法
最后需要关注的问题是如何将模拟得到的能量和能垒转化为实验上可以测量的电流密度和法拉第效率。衔接微观模拟结果和宏观可观测量的桥梁是动力学蒙特卡洛方法。简单来说,可以将动力学蒙特卡洛方法看成是一种微观模拟的粗粒化方法。也就说,将具体的反应和反应过程简化为一个通过速率描述的事件,并且这些事件是相互关联的。这些事件发生的相互顺序可以通过蒙特卡洛规则进行选取。最终实现反应过程的全模拟。
动力学蒙特卡洛需要的主要输入可以通过量子力学计算获得。所以,预测的准确性直接取决于量子力学计算的准确性。而且,反应速率与能量之间是指数关系,能量预测的微小偏差可以直接导致巨大的预测误差。这也是动力学模拟在大多数情况下,无法与实验数据进行直接比对的原因。
总结一下,在电化学理论模拟中,有多种流行的计算方法和模拟策略。通常来说,理论模拟方法越高级、模型越接近实验条件,那么准确性会更高。当然,在模拟中如果能抓住问题的关键,也可以在合适的计算方案下取得合理的计算结果。在电化学模拟中,目前最为完备、近似最好的模拟方案是:PBE-D3方法+杂化模型+自由能计算组合。理论上来说,上述方法的准确性应该是最高的,相应的计算量也比较昂贵。对于一般的计算,或者以高通量筛选为目标的理论预测,隐式水分子模型的静态计算也能获得不错的计算结果。综上所述,我们推荐的模拟策略是:高精度计算研究反应机理+高效率计算进行材料扫描。
3 反应机理的理论研究
对于二氧化碳产物反应路径的研究一直是人们关注的研究热点。对于反应机理的深入理解是进行催化剂理性设计的前提条件。尤其是对于像二氧化碳还原这类复杂的化学反应。举例来说,二氧化碳还原制备乙烯涉及到12个电子转移。没有指向性的催化剂合成存在效率低下的致命缺陷。所以,理解反应机理是非常重要的。
以下我们将对二氧化碳还原产物反应机理的研究的主要进展进行介绍。
3.1 一氧化碳的生成路径
一氧化碳是一种特殊的二氧化碳还原产物。因为,它同时也是通向深度还原产物的反应中间体。由于其高度的稳定性,一氧化碳是唯一可以完全确认的反应中间体。研究发现,深度还原产物一定要经过一氧化碳,并且其反应过电位决定了整个反应的过电位。所以,对于二氧化碳还原的研究可以分为两个阶段:第一个阶段是二氧化碳还原为一氧化碳;第二个阶段是一氧化碳到其它深度还原产物。
虽然二氧化碳还原生成一氧化碳的研究早有理论研究报道,但是其关键反应中间体依然存在争议。Peterson 等人报道了通过*COOH中间体得到一氧化碳的反应中间体42。理论预测的还原电位为-0.41 V (RHE)。作者并没有对反应能垒进行预测。但是,大量的实验研究表明,在形成*COOH中间体的同时可能存在另一个更为重要的反应中间体,并且形成这个中间体的路径可能是真正的决速步骤。利用分子动力学显式溶剂方法,我们成功的得到了*COO-这个反应中间体(如图5所示)。并且证实了该中间体为二氧化碳还原生成一氧化碳的反应决速步骤。通过分析模拟结果,我们发现,在模拟中获得该中间体存在两个必需的条件:首先,稳定该中间体需要至少两个氢键。这一点通过显式溶剂模拟可以实现,而真空模拟和隐式溶剂模拟都存在一定的困难。其次,稳定该中间体需要合适的电位。在二氧化碳还原电位下,该中间体承载一部分负电荷,这部分负电荷是形成共轭结构的关键,这一点通过在模拟中施加负电压实现。上述分析结果,一方面解释了以往模拟无法获得*COO-中间体的原因;另一方面也再次强调了在模拟中充分考虑真实反应环境的必要性。
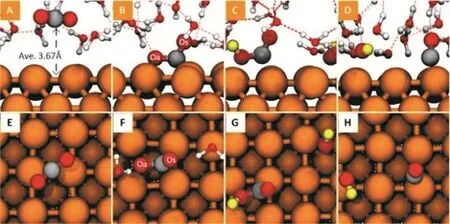
图5 从二氧化碳还原产生一氧化碳的反应路径45Fig. 5 The reaction pathway of CO formation from CO2.
该反应中间体后续得到了实验光谱的证实。在模拟中,为了确保*COO-是反应中间体而不是瞬态过渡态,我们将模拟扩展到40 ps。在整个模拟时间内,*COO-都可以稳定存在。并且在模拟中,我们可以进一步确认*COO-的吸附构型,这对解释实验光谱具有重要作用。
3.2 甲酸的生成路径
甲酸(或甲酸盐)是另一种两电子还原产物。与一氧化碳类似,在合适的催化条件下可得到很高的选择性。实验证据显示,甲酸的生成与一氧化碳的生成是两种截然不同的反应路径。根据理论计算,HCOO*被认为是生成甲酸的反应中间体43,44。尽管尚无直接的实验证据证明其正确性,但是上述机理是目前应用最为广泛的甲酸生成机理。
我们的理论计算发现了不同于已有报道的反应机制45。在我们反应机理中,甲酸阴离子可以通过Eley-Rideal (ER)反应直接获得。具体来说,该反应的反应物为表面吸附H和液体中的CO2,通过两个电子转移直接实现甲酸阴离子的生成。该反应机理可以合理地同时解释两个关键的实验数据:第一甲酸的高选择性46,第二甲酸与HER的竞争机制47。上述推理基于两个原因:首先,基于ER机理的HCOO-形成机理,仅需要一步即可实现。效率要远高于传统上一般认为的两步反应。这可以解释甲酸的高选择性。其次,基于ER机理的HCOO-形成机理,需要表面吸附的H原子。而HER也需要表面吸附的H原子才能够进行。因此,HCOO-的形成构成了与HER的直接竞争。这和实验上观测到的结果是吻合的。
由于尚缺乏直接的实验证据,尽管我们认为直接机理更为合理,但是,目前甲酸的形成尚无统一的认识。
3.3 甲烷的生成路径
甲烷是Cu还原二氧化碳的主要碳氢产物之一(另一种是乙烯)48。甲烷的形成与Cu的晶面有明显关联。研究表明,Cu(111)晶面有利于甲烷的形成。根据理论计算,人们提出了甲烷的反应机理49。其反应步骤包括:CO2,*COOH,*CHO,*CH2O,*OCH3,最后是CH4。需要注意的是,上述反应机理是基于真空计算的计算结果获得的。所以,部分中间体存在明显的不合理性。例如,*OCH3这个反应中间体很难在液相反应中得到。
我们的理论计算发现了一条完全不同的反应机制。在我们的反应机理中,甲烷的形成机理是CO → *CHO → *CHOH → *CH → *CH2→ *CH3→ CH4(如图6所示)。其中,*CHO的生成同时是反应的热力学和动力学决定步骤。溶剂化效应可以明显降低*CHO的反应能垒。在考虑显式溶剂作用的情况下,反应能垒从真空计算得到的0.96 eV,降低到0.55 eV。于此同时,对于含有OH基团的反应中间体(如*CHOH),溶剂化(尤其是显式溶剂化模型)也提供了更强的稳定作用,一般可以达到0.1 eV。
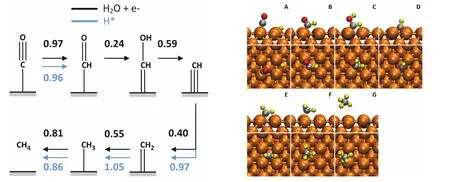
图6 从一氧化碳还原产生甲烷的反应路径51Fig. 6 The reaction pathway of methane formation.
甲烷生成也依赖于催化剂晶面。上述*CHO中间体适用于Cu(100)晶面。对于Cu(111)晶面,研究显示,*COH的形成更有可能是反应的热力学和动力学决定步骤。在pH = 1的情况下其反应能垒0.8 eV。当然,*COH也会被还原为*CHOH,后续的反应路径与在Cu(100)上的反应路径完全相同。
虽然,甲烷生成的决速步骤在CHO和COH两者之间依然存在一定的争议,但是*CHOH中间体应该是必经的反应中间体。目前,通过*CHOH形成甲烷的反应路径是当前应用最为广泛的反应机理。
3.4 乙烯的生成路径
乙烯是Cu还原二氧化碳的另一种主要碳氢产物48,50。与甲烷类似,乙烯的生成也具有晶面的选择性。研究表明,Cu(100)晶面有利于乙烯的生成。研究乙烯的反应过程首先要解决C―C键的形成。早期,人们对于C―C键形成提出了多种可能的设想。随着实验研究的推进,人们发现乙烯的形成没有SHE的依赖性52。也就是说C―C键的形成过程中,不涉及到质子。所以,通过两个CO耦连的方式形成C―C键是唯一的可能性。CO―CO反应过程虽然不涉及到质子,但是存在电子转移。本质上依旧是电化学反应,其电化学反应方程式为:

根据巨正则系综电子化学势模拟可以推导出δ在0.5左右,符合传统电化学的理论预期。进一步理论计算表明,该反应的反应能垒是乙烯路径的决速步骤。理论模拟预测CO耦连的反应能垒在0.69 eV左右(如图7所示),这也与实验观测到的起始电位吻合。
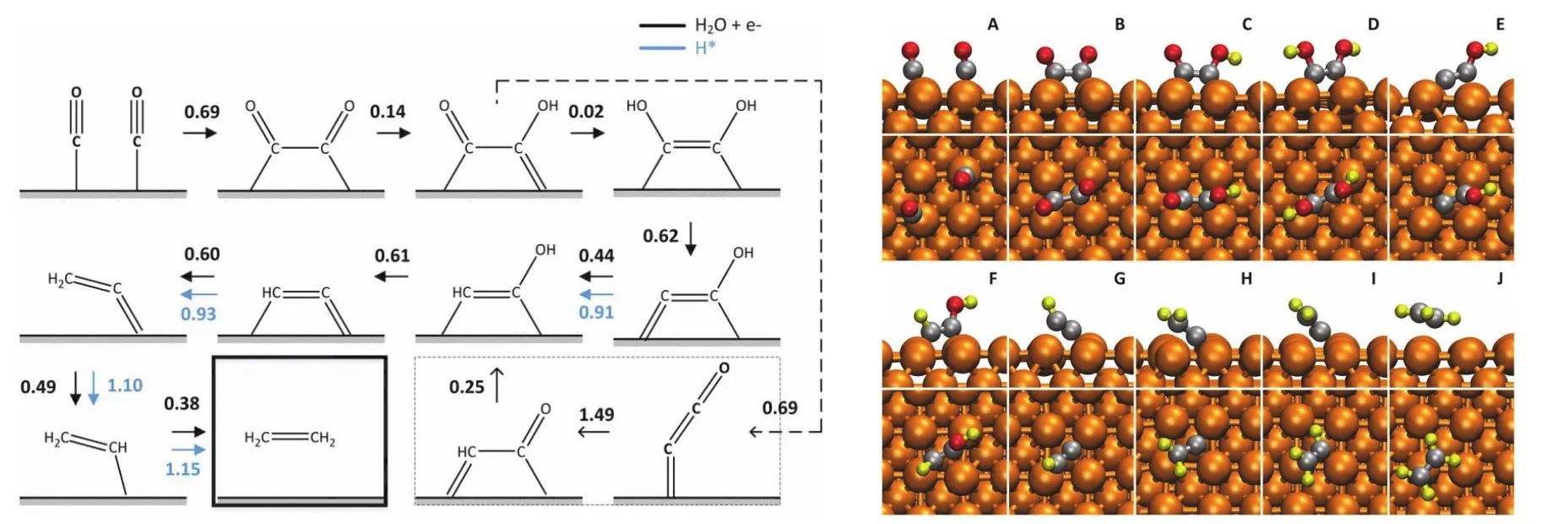
图7 从一氧化碳还原产生乙烯的反应路径51Fig. 7 The reaction pathway of C2H4 formation.
从上述的分析可以发现,CO耦连最有可能是生成乙烯的初始反应53。用上述准则可以剔除掉一些不合理的反应54。此外,甲烷和乙烯存在着竞争机制。如果CO形成*CHO更为容易,那么将得到C1产物,这是Cu(111)的情况。反之,如果CO耦连更为容易,那么将得到C2产物,这是Cu(100)的情况。
关于C―C键的形成,也存在其他的解释方式。例如在Ma等人的工作中,作者指出C―C键可能是在H的协助下发生的,即先形成*CHO然后再进行C―C耦合55。
尽管乙烯生成的起始反应已经基本确定,但是在CO耦连后的反应步骤依然存在争议。人们基于不同的理论预测方法,提出了不同的假设。目前并没有任何确凿的实验证据对上述机理进行验证。
3.5 乙醇的生成路径
在Cu(111)和Cu(100)表面上,乙醇的产量要低于乙烯。但是,在一些特定的催化体系中,乙醇的产量很有可能高于乙烯。从实验结果来看,乙醇和乙烯应该都是从一个共同的反应中间体而来。这个共同的反应中间体,可能是*OC―CO,也可能是*OC―CO后的某一个反应中间体。但是,实验上目前无法确定这个共同反应中间体的具体结构。在实际反应过程中,乙醇可能通过多种反应路径得到。这也增加了理论预测的难度。虽然,有实验证据表明,乙醛可以还原为乙醇。但是,这并不能作为乙醛是乙醇中间体的证据。总的来说,乙醇的详细生成路径尚未统一。
3.6 乙酸的生成路径
乙酸是CO还原中非常特别的产物。原则上,在CO还原中生成乙酸需要引入一个*O,这是一个氧化反应的特征。但是这显然违背了反应处于还原环境这一前提。我们认为,和其他反应物不同,乙酸的生成一定和表面吸附的物种,如氧或*OH相关。因此,我们提出了表面*O参与的反应机制。该反应机理可将*O加成有效的转化为电化学还原反应。但是,值得注意的是,上述反应机理依赖于表面吸附*O的稳定性。
另一种乙酸生成机制是Luc等人提出的,其关键步骤在于CH2CO水解生成乙酸和乙醇56。上述反应虽然能够一定程度的解释实验现象,但是也存在明显不足:首先,CH2CO水解生成乙酸的同时,必然伴随着乙醇的生成。这和实验中观测到的乙酸单一产物不符。其次,反应过程中需要CH2CO的表面脱除,这也不太符合表面反应的要求。总之,乙酸的生成路径尚无统一认识。
3.7 甲醇的生成路径
甲醇在CO2还原产物中的比例非常低。其反应机理相对简单。甲醇和甲烷在反应前期具有相同的反应路径。两种反应在*CHOH中间体后开始出现差别。对于甲醇来说,保持OH进行两步加氢反应即可实现甲醇的生成。尽管反应路径比较清晰,但是在电化学还原条件下,理论计算表明OH脱除非常容易。这也合理解释了实验中甲醇产量非常低的内在原因57。根据有已有文献报道,Nitopi等人对已有报道的反应机理进行了总结,如图8所示。
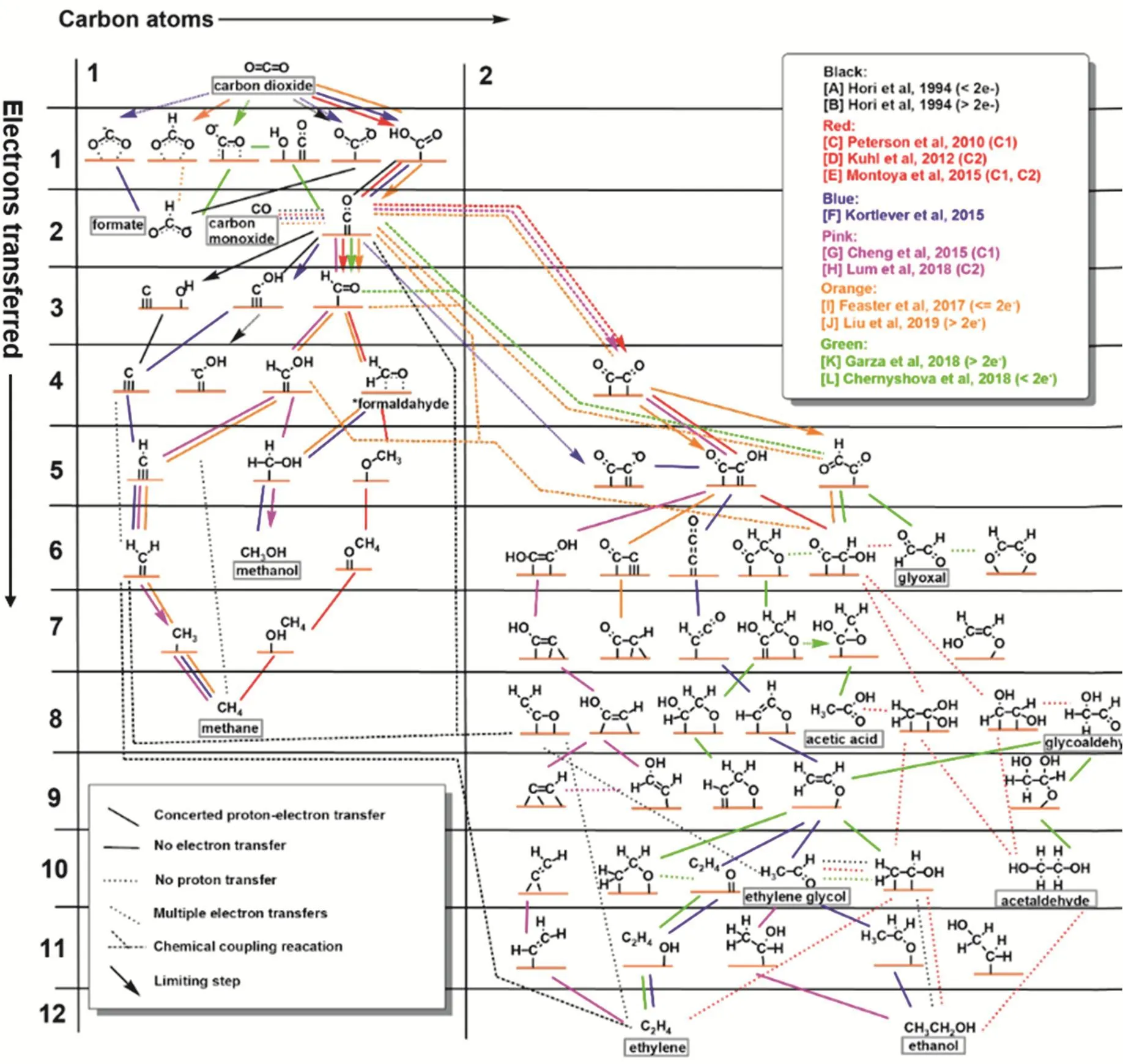
图8 在多晶铜上将二氧化碳还原为C1和C2产物的可能机理8Fig. 8 Possible mechanistic pathways of CO2 reduction to C1 and C2 products on polycrystalline copper.
当然,CO2RR还可能存在多种可能的方式。例如草酸盐产物很有可能是CO2直接耦合而形成的58。本文受篇幅所限,不一一介绍。建议参考其他综述文章59,60。
4 反应中间体的确认
由于计算中采用的计算方法不同,不同的理论计算往往得出不同的预测结果。那么,如何在众多可能的反应机理中,甄别出最为合理的反应路径。目前最为可行的方法就是利用理论与实验相结合的方法对反应中间体和反应机理进行验证61。以下介绍几种在电化学研究中常用的实验方法。
4.1 同位素原子示踪法
同位素原子示踪法是一种研究化学反应机理的有效手段。利用同位素标定,可以有效的追踪原子从反应物到产物的转移过程,从而确认反应机理。同位素原子示踪法是目前确定反应中元素迁移过程最好的技术手段。在二氧化碳电还原的研究中,同位素原子示踪法也是一种行之有效的技术手段62,63。
4.2 常压X射线光电子能谱
X射线光电子能谱(简称XPS)为电子材料研究提供了各种化合物的元素组成和含量、化学状态、分子结构、化学键等方面的信息。与传统的在超真空条件下工作的XPS技术不同,常压光电子能谱(AP-XPS)可以研究催化剂颗粒在较高温度下、一定压力(133.32 Pa左右)气体环境中的表面化学。近十年来,AP-XPS广泛用于研究多种催化剂在反应条件下的表面化学,包括单晶模型催化剂、金属颗粒催化剂、金属氧化物与碳化物催化剂。在二氧化碳电还原的研究中,AP-XPS也是一种非常有效的研究手段。理论模拟可以实现XPS的预测,并且具有可以和实验数据相比拟的精度。因此,理论预测结合实验结果是辨别电化学界面结构和反应中间体的有效手段64,65。
4.3 表面增强红外和表面增强拉曼光谱
表面增强红外和表面增强拉曼光谱是研究表面吸附分子振动模式的有效手段。分子振动光谱是合成化学中最常用的表征手段。原则上,分子振动光谱也是电化学研究中的有力工具。但是,由于表面吸附情况复杂,尤其是在电解液存在的情况下,表面物种的特征峰容易被电解液的特征峰重叠。这种情况下,具有更高精确度的表面增强红外和表面增强拉曼光谱能够更好的发挥作用66-69。理论预测与实验相结合可以更有效的辨别反应中间体(如图9所示)8,70,71。

图9 反应中间体的结构和预测得到的振动态密度78Fig. 9 The snapshots of the reactive intermediates and the predicted vibrational density of states.
5 二氧化碳合成的优化策略
5.1 纳米催化
虽然体相Cu的催化性能有限,但是Cu的纳米颗粒却显示出了更为优越的催化性能。因此,通过将金属进行纳米调控,可以实现大幅度提升催化性能的目的。与此同时,由于纳米化导致的电子结构改变,催化剂纳米化也会对产物的选择性造成影响。纳米金属和纳米合金是目前针对二氧化碳电还原催化效果最好的体系72-75。
5.2 表面功能化
另一种提高催化性能的手段是表面功能化。例如,通过引入功能化分子、表面活性剂或者聚合物。虽然并没有对催化剂本身进行改变,但是实验结果显示,功能化也能大幅度提升材料的催化性能。与此同时,稳定性也能得到大幅度提升76,77。从工业应用的角度,表面功能化是非常行之有效的优化策略。
6 讨论和展望
6.1 新型成键方式
虽然,二氧化碳电还原两电子产物目前已经可以优化到非常高的选择性,但是二氧化碳电还原深度产物更具有综合经济价值。所以,通过调控催化剂优化深度产物的生成路线是收益更高的目标。二氧化碳电还原深度产物涉及到多个电子转移过程,这从根本上限制了产物的产率。举例来说,从二氧化碳到乙烯涉及到12个电子转移,即使每一步的可以达到90%的效率,经过12步反应后能达到的效率不到30%。因此,在优化催化剂的同时,更为有效的方法是改进二氧化碳的形成路线。在合成复杂产物的同时,尽量简化合成路线。为了实现上述目标,需要我们打破常规的合成思路,探索新的成键方式。例如,我们可以跳出传统CO (或者CO2)的合成框架,通过引入其他分子,丰富产物的同时,简化合成步骤。例如,Jouny等人报道了通过在CO还原中引入NH3的方法合成乙酰胺的研究工作。在这个工作中,NH3的引入实现了通过C―N耦合的新型合成路线,于此同时也将合成路线减少了两个电子转移步骤,有利于提高产率。具体反应过程如如图10所示79。此外,Chen等人通过在CO2还原中引入N2,实现了尿素的电化学合成80。这个工作值得称道之处包括:提出了一种新型的CO插入N2的反应机理,同时实现了CO2还原和N2还原这两个重要的电化学反应。Feng等人也报道了类似的反应80。
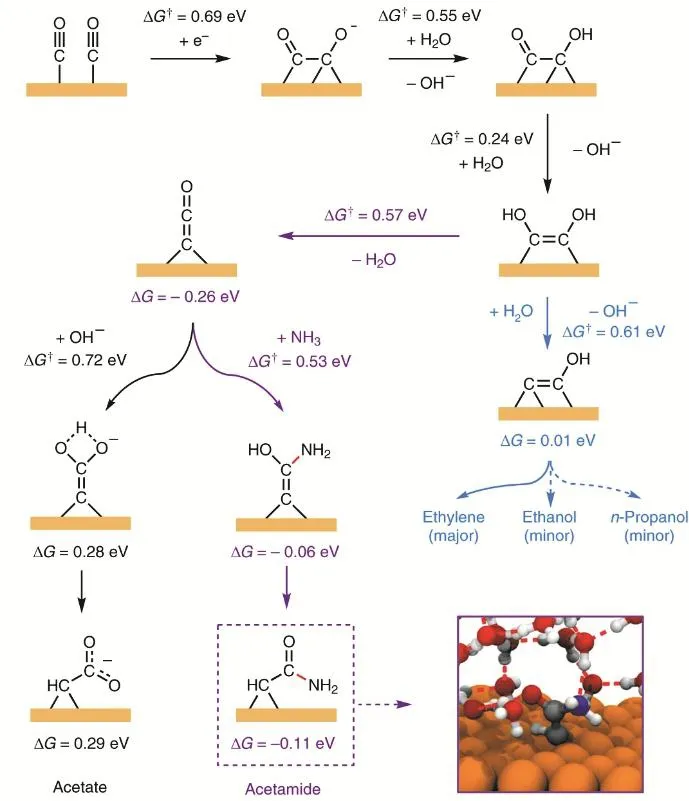
图10 碳-氮耦连的反应路径79Fig. 10 The reaction pathway of C―N coupling.
6.2 机器学习在二氧化碳电还原中的应用
近些年来,机器学习是电化学理论研究中的一个热门研究方向。机器学习是人工智能的一个分支。因为机器学习算法中涉及了大量的统计学理论,机器学习与推断统计学联系尤为密切,也被称为统计学习理论。机器学习的基本思想是对已有数据进行训练,产生的预测模型用于新数据的预测。
在电化学催化领域,机器学习的潜在应用有很多。举例来说,目前反应机理的研究建立在量子化学计算的基础上。虽然,量子化学方法的计算精度很高,但是计算量过于昂贵。所以,如果利用已有量化数据,通过训练产生可靠的预测模拟,就可以大幅度提高预测效率。从而加速机理研究和材料开发。例如,Ma等人利用CO吸附能为描述符,利用机器学习预测了多种金属的CO2还原性能81。Tran和Ulissi进一步结合HER预测,对多种金属间化合物的CO2还原性能进行了理论预测82。在机器学习预测的帮助下,Zhong等人实验合成了CuAl合金,达到了高效的CO2还原制备C2H4的目的(如图11所示)83。
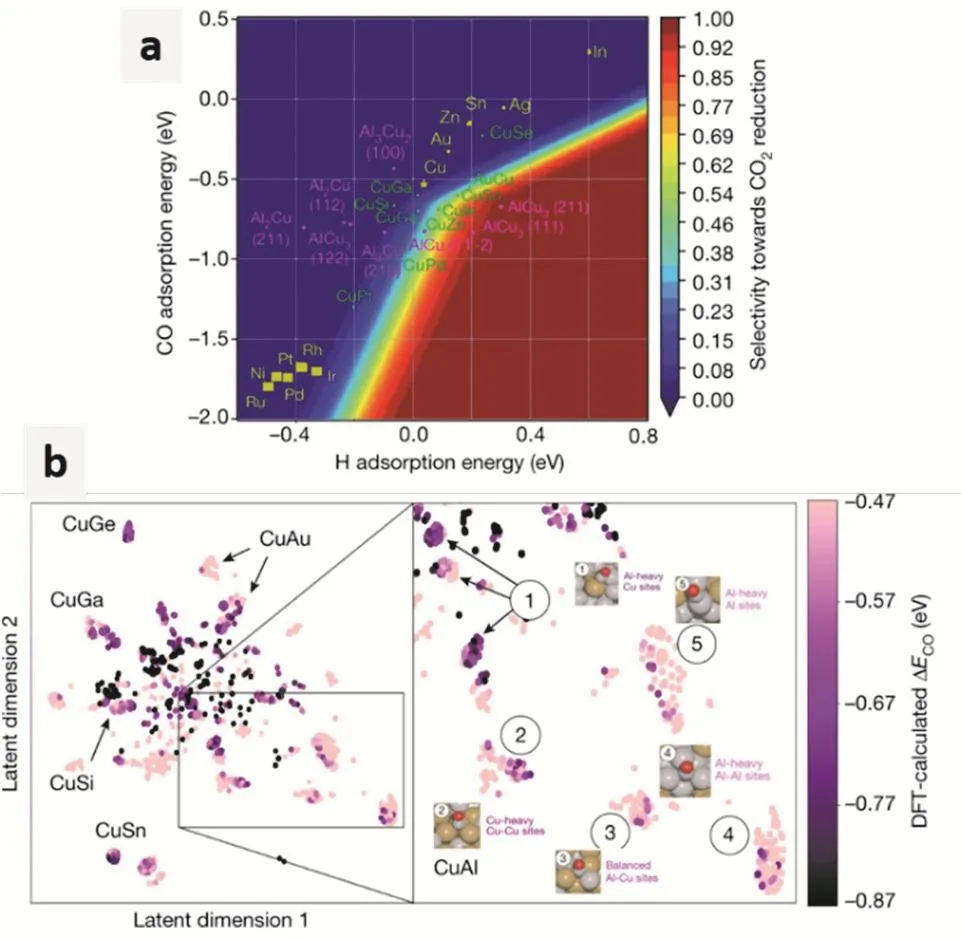
图11 使用计算方法筛选铜和基于铜的化合物83Fig. 11 Screening of Cu and Cu based compounds using computational methods.
机器学习方法之所以能够发挥作用,是因为高质量的机器学习方法能够给出化学精度的预测结果,从而保证预测质量。传统的经验模拟已经在预测领域发挥了巨大的作用。但是,受限于模型的局限性,其精度往往很难系统性地提升。而先进的机器学习方法,如深度学习(或深度神经网络等)为拟合复杂的化学空间提供了可能。在二氧化碳电还原的研究中,机器学习正发挥着越来越重要的作用81,82,84。
当然,深度学习也有其自身的问题,预测精度的提升往往需要复杂的神经网络,这也就意味着更多的参数。为了拟合这些参数,对数据集的数据量提出了更高的要求。此外由于过拟合而造成的错误预测,也是深度学习中必须要解决的问题。
7 总结
在本工作中,我们系统地总结了近些年来在二氧化碳电还原领域的理论研究进展和成果。其中重点关注了反应机理的研究。因为,对于二氧化碳电还原这一重要的研究课题来说,其复杂的反应网络造成了机理研究中的困难和挑战。而对于反应机理认识的缺失,直接限制了催化性能的进一步提升。
理论模拟为理解电化学反应过程和反应机理提供了有力的研究手段。随着计算机硬件水平的不断提升和理论模拟方法的完善。理论预测的精度不断提高。在电化学这类反应条件复杂、实验表征困难的研究体系中,理论模拟方法正发挥着越来越重要的作用。值得注意的是,理论模拟中存在着不同程度的近似,在理解和解读预测结果时,需要首先将方法自身的准确性考虑在内。一般来说,如果计算方法越精确、计算模拟越接近实验条件,那么其预测结果也更为准确。相应的计算量也越昂贵。当然,如果模拟中能够抓住物理本质,在较小的计算量下也能得到不错的预测结果。
近些年来,通过理论结合实验,人们对二氧化碳电还原各种产物反应机理的认识日趋完整。其中,甲烷的形成机理已经基本确定。乙烯的初始C―C键形成过程也得到了理论和实验的确认。除此之外,其他多种产物的最优反应路径也通过多种理论预测建立起来。随着理论计算方法的发展和原位表征手段的提高,将会有越来越多的反应中间体被检测到,从而对反应机理进行确认。
对于反应机理的认识有利于加速催化剂设计。通过纳米调控和表面改性等方法,大量具有良好性质的铜基催化剂已经完成实验合成,这些具有高活性、高选择性和高稳定性的先进功能材料为打开二氧化碳电还原工业化道路打下了良好的基础。二氧化碳电还原深度还原产物的形成路线涉及到多个电子转移和多个反应中间体,这是其活性和选择性难以进一步提高的根本制约因素。为了打破上述限制,需要我们跳出已有的合成思路,通过提出新颖的成键方式简化合成步骤,在提高活性和产率的同时丰富二氧化碳电还原产物。
我们可能处于一个机器学习和人工智能走向大面积应用的新时代。这些新的技术手段将对理论模拟乃至整个化学领域带来深远的影响。随着计算方法的日趋成熟,人工智能和大数据驱动的机理研究和材料设计已经表现出了无尽的潜力。这些优质、高效的理论新工具将大幅度加速理论研究和材料开发。
猜你喜欢
哈哈画报(2022年8期)2022-11-23 06:21:32
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
建材发展导向(2021年14期)2021-08-23 00:57:14
小学科学(学生版)(2021年5期)2021-07-22 02:40:08
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
学生天地(2020年18期)2020-08-25 09:29:24
中国煤层气(2019年2期)2019-08-27 00:59:30
环境与可持续发展(2017年2期)2017-04-06 03:07:30
汽车零部件(2014年8期)2014-12-28 02:03:03

- 物理化学学报的其它文章
- Understanding the Role of Cu/ZnO lnteraction in CO2 Hydrogenation to Methanol
- Electrocatalytic CO2 Reduction to Ethylene over CeO2-Supported Cu Nanoparticles: Effect of Exposed Facets of CeO2
- Controlling the Global Mean Temperature by Decarbonization
- Cu@UiO-66 Derived Cu+-ZrO2 Interfacial Sites for Efficient CO2 Hydrogenation to Methanol
- 离子液体介导CO2化学转化研究进展
- 过渡金属催化CO2/H2参与的羰基化研究进展