
牛DHX36解旋酶及其突变体底物结合与解旋活性研究
2021-06-01 03:06:12郭青青郭海磊刘娜女奚绪光
西北农林科技大学学报(自然科学版) 2021年5期
郭青青,郭海磊,刘娜女,奚绪光
(西北农林科技大学 生命科学学院,陕西 杨凌 712100)
解旋酶是一类可以水解三磷酸腺苷释放能量并打开核酸之间氢键的一类分子“马达”蛋白,广泛存在于病毒、细菌和真核生物中[1-2],参与细胞的多种代谢过程,如DNA的复制、转录、翻译及RNA剪切等生命活动[3-5]。编码解旋酶的基因一旦发生突变或缺失,将无法表达相应的蛋白,进而引起多种严重的遗传性疾病,如Bloom综合症、Werner综合症、Rothmund-Thomson综合症等[6-8]。根据解旋酶的序列保守性、结构相似性及解旋方向性,可以将其分为6个超家族:SF1、SF2、SF3、SF4、SF5、SF6[9]。SF1和SF2是其中最大的两个超家族,SF1是目前研究得最清楚的超家族;SF2是含有众多亚家族的超家族,其中研究较多的是DEAD-box、DEAH/RHA和Snf2等亚家族[10-12]。
富含鸟嘌呤的核酸序列可以通过4个鸟嘌呤自行组装成四链体结构,即G4结构。生物信息学分析发现,能够形成G4结构的基因序列多存在于特定的基因组区域,例如端粒、核糖体DNA、转录因子结合位点、启动子区域[13-15]等。由于位置的特殊性,G4结构可能参与了基因表达调控的许多生物学过程[16-17]。G4结构主要有两类,即DNA G4结构和RNA G4结构。有研究发现,人类细胞中的DNA G4结构在DNA复制、修复以及维持染色体的稳定等方面起着重要的作用。在DNA复制过程中,DNA G4结构的出现会阻碍复制的顺利进行,此时就需要解旋酶来进行修复[18]。真核细胞中有许多解旋酶,其中不少参与G4结构的解旋活动[19-20]。
DHX36(也称为RHAU或G4R1)是SF2超家族中DEAH/RHA亚家族的成员,是对含有G4结构的底物具有较高亲和力的一类ATP依赖型RNA解旋酶[19-21]。DHX36的基本组成是解旋酶核心区域以及位于两侧的N端和包含OB域的C端区域。目前,人们对DHX36 N端区域的研究较为深入,根据生物信息学分析,将其分为两个部分:富含甘氨酸区和RSM区[22]。Lattman等[23]发现,DHX36 N端的RSM区是参与G4结构结合和解旋活动必不可少的部分,截去N端区域后蛋白不具有解旋活性。还有研究表明,DHX36的RSM区氨基酸序列能够识别端粒上的G4结构并参与端粒延伸的调控[24]。说明DHX36的N端区域是参与G4结构解旋的重要部位。据报道,DHX36能够与G4结构相互作用,从而对转录过程进行调控,其还在小鼠心脏发育和造血过程中发挥重要作用[25-26]。Chen等[27]研究发现,果蝇DHX36(DmDHX36)N端的RSM区和C端的OB域是参与G4结构结合及解旋的主要部位。
目前,因人DHX36(hDHX36)蛋白在大肠杆菌表达系统中表达难度较大,因此有关hDHX36的酶学特征和结构研究相对较少。本研究选择与hDHX36同源性较高且可在原核表达系统中表达的牛(Bostaurus)DHX36(BtDHX36)解旋酶为材料,利用原核表达系统获得高纯度野生型BtDHX36蛋白、RSM区突变体蛋白(BtDHX36R63AI65A、BtDHX36Y69A、BtDHX36K76AN77AK78A)和OB域突变体蛋白(BtDHX36Y862A),利用荧光各向异性法(FA)和快速停流-荧光共振能量转移(FRET)技术,比较野生型BtDHX36及其突变体对底物结合和解旋活性的差异,旨在为牛DHX36的酶学特征和结构研究提供参考。
1 材料与方法
1.1 材 料
1.1.1 质粒、载体及菌种 野生型BtDHX36蛋白编码的基因序列(载体PBHM-BtDHX36),由Bio-Matik公司合成;表达载体pET15b、大肠杆菌菌株Top10、BL21(DE3)和质粒pET15b-sumo-DmDHX36均由西北农林科技大学生命科学学院生物大分子实验室保存。
1.1.2 主要试剂、柱材及仪器 T4 DNA ligase、Prime STAR DNA polymerase,购自Takara公司;EcoRⅠ、XhoⅠ、NdeⅠ等限制性内切酶,购自NEB公司;Ni-NTA Beads、Hi Trap SP HP预装柱,购自GE healthcare公司;低温超高压破碎仪,购自JNBIO公司;超声波破碎仪,购自宁波江南仪器厂;AKTA purifier蛋白纯化仪,购自GE healthcare公司。
1.2 方 法
1.2.1 DNA底物的准备 根据文献[28],设计用于DNA结合试验的底物16nt-ssDNA、Telomere G4和用于解旋试验的底物Telomere G4-16T序列,交给生工生物工程(上海)股份有限公司合成,其中16nt-ssDNA和Telomere G4底物均标记荧光素(fluorescein,F),Telomere G4-16T底物标记荧光素(fluorescein,F)和六氯荧光素(hexachloroflourescein,HF),底物序列见表1。16nt-ssDNA底物用ddH2O稀释后备用;Telomere G4和Telomere G4-16T底物分别置于退火缓冲液(100 mmol/L KCl,20 mmol/L Tris-HCl pH=7.5)中,100 ℃加热使其变性5 min,然后缓慢冷却至室温,备用。
1.2.2 野生型BtDHX36及其突变体表达载体的构建 野生型BtDHX36及其突变体表达载体构建流程如图1所示。将含有BtDHX36基因的载体PBHM-BtDHX36用EcoR Ⅰ和XhoⅠ双酶切,得到野生型BtDHX36基因片段。使用实验室已构建的pET15b-sumo-DmDHX36质粒为模板,以sumo-F为上游引物(序列:5′-GCCATATGAGCGATAGCGAAGT-3′),sumo-R为下游引物(序列:5′-CAGATTGGCGGCGAATTCCA-3′),扩增具有促进蛋白溶解作用的sumo基因片段,PCR扩增体系为50 μL:5×Prime STAR Buffer(Mg2+plus)10 μL、Prime STAR DNA polymerase(2.5 U/μL)0.5 μL、dNTP Mixture(2.5 mmol/L)4.0 μL、sumo-F和sumo-R(10 μmol/L)各1 μL、模板(30 ng/μL)1 μL、ddH2O 32.5 μL;PCR反应程序为:98 ℃预变性3 min;98 ℃变性15 s,60 ℃退火15 s,72 ℃延伸30 s,共30个循环;72 ℃终延伸7 min后,16 ℃保温。对sumo基因片段进行NdeⅠ和EcoRⅠ双酶切,使用NdeⅠ和XhoⅠ双酶切pET15b载体。将BtDHX36、sumo基因片段、pET15b按照3∶3∶1物质的量比加入连接体系,16 ℃连接过夜,将连接产物转入Top10克隆菌,涂至平板后于37 ℃培养8 h,对菌落进行PCR、质粒EcoRⅠ和XhoⅠ双酶切鉴定,最终获得野生型重组质粒pET15b-sumo-BtDHX36。
采用Overlap PCR 在BtDHX36中引入点突变,用突变后的BtDHX36基因片段构建4种BtDHX36突变体(mutant)(RSM区突变体BtDHX36R63AI65A、BtDHX36Y69A、BtDHX36K76AN77AK78A和OB域突变体BtDHX36Y862A)的重组质粒,并进行菌落PCR、质粒EcoRⅠ和XhoⅠ双酶切鉴定,具体方法同上。
1.2.3 野生型BtDHX36及其突变体的诱导表达与纯化 将鉴定正确的野生型重组质粒pET15b-sumo-BtDHX36转化到大肠杆菌BL21(DE3)感受态细胞中。挑选单克隆进行活化,37 ℃扩大培养,当菌液在600 nm处的吸光度(OD600)为0.7~0.8时,加入IPTG(终浓度0.3 mmol/L),18 ℃诱导14 h后取样。将样品进行4 500 r/min离心(10 min),收集菌体沉淀,按质量(g)体积(mL)比1∶10加入裂解Buffer(20 mmol/L Tris-HCl pH=7.9,500 mmol/L NaCl,体积分数5%甘油,5 mmol/L咪唑)复溶,先利用低温超高压破碎仪破菌4次,再使用超声波打断DNA双链以降低溶液黏度,然后于12 000 r/min离心45 min,分别留取部分上清液和沉淀样品。用0.45 μm滤膜过滤上清液,将其载入Ni-NTA亲和层析柱,使用Elute Buffer(20 mmol/L Tris-HCl pH=7.9,500 mmol/L NaCl,体积分数10%甘油,300 mmol/L咪唑)洗脱,留取部分洗脱样品。将含有目的蛋白的洗脱样品按1∶1 000的物质的量比加入sumo酶,4 ℃酶切过夜,留取部分酶切样品。用稀释Buffer(20 mmol/L Tris-HCl,pH=7.9,体积分数10%甘油)稀释蛋白样品至NaCl终浓度为80 mmol/L,然后使用Hi Trap SP HP柱梯度洗脱,将得到的洗脱样品进行10%的SDS-PAGE电泳检测,合并较纯组分并用30 ku的Millipore超滤管离心浓缩,分装后立即用液氮速冻,置于-80 ℃保存。对上述各步所得样品进行10%的SDS-PAGE电泳检测。同法对BtDHX36突变体蛋白进行诱导表达与纯化。
1.2.4 野生型BtDHX36体外最佳底物结合条件试验 以16nt-ssDNA为底物,检测野生型BtDHX36与底物的体外最佳结合条件,考察因素包括KCl、MgCl2浓度、反应温度、pH,反应体系150 μL,其中含5 nmol/L荧光标记底物的16nt-ssDNA、野生型BtDHX36蛋白(0,4,8,12,16,20,24,36,50,100,200,300 nmol/L)和Buffer(20 mmol/L KCl、2 mmol/L MgCl2、20 mmol/L Tris-HCl,pH=7.5)。分别在不同的KCl浓度(0,50和100 mmol/L)、反应温度(25,30和37 ℃)、MgCl2浓度(0,2和5 mmol/L)、pH(6.5,7.5和8.5)下孵育5 min,用Infinite F200/M200型多功能酶标仪(瑞士Tecan公司)测定每种反应条件下的各向异性值(Δr),每次试验重复3次。用Origin软件拟合蛋白浓度与Δr的曲线,由此曲线获得理论最大各向异性值(Δrmax),用希尔方程Δr=Δrmax×P/(Kd+P),计算平衡解离常数(Kd),式中P为解旋酶的浓度。以Kd为判定指标,确定野生型BtDHX36蛋白与16nt-ssDNA的体外最佳结合条件。
1.2.5 野生型BtDHX36及其突变体的底物结合活性差异分析 在最佳底物结合条件下,以Telomere G4为底物,检测野生型BtDHX36及其4种突变体(BtDHX36R63AI65A、BtDHX36Y69A、BtDHX36K76AN77AK78A、BtDHX36Y862A)蛋白与底物结合活性的差异。具体操作为:使用酶标板加样,结合反应体系为150 μL/孔,其中含5 nmol/L底物和不同浓度(0,5,10,15,20,30,40,60,100,200,400 nmol/L)的上述5种蛋白,振荡混匀,37 ℃孵育5 min,酶标仪检测得到其对应的Δr,每次试验重复3次。用Origin软件拟合蛋白浓度与Δr的曲线,由此曲线获得Δrmax,进而得到结合比例(Δr/Δrmax), 用Origin软件拟合蛋白浓度与结合比例的曲线。使用希尔方程计算Kd,比较5种蛋白结合活性的差异。
1.2.6 野生型BtDHX36及其突变体的解旋活性差异分析 利用Bio-Logic SFM-400停流装置结合荧光共振能量转移(FRET)原理,以Telomere G4-16T为底物,测定得到野生型BtDHX36及其突变体蛋白的解旋动力学曲线,并以速率常数为指标分析野生型BtDHX36及突变体蛋白的解旋活性差异。试验重复3次,所有解旋数据均参考Zhang等[29]的方法进行分析。
2 结果与分析
2.1 野生型BtDHX36及其突变体的载体构建与蛋白的诱导表达和纯化
2.1.1 野生型BtDHX36表达载体的鉴定及蛋白纯化 野生型BtDHX36经菌落PCR鉴定,得到2.8 kb的片段(图2-A),与预期结果一致。挑选阳性菌落,提取质粒,将重组质粒pET15b-sumo-BtDHX36经EcoRⅠ和XhoⅠ双酶切后,出现了约2.8 kb的目的基因片段(图2-B)。将重组质粒pET15b-sumo-BtDHX36转入大肠杆菌BL21(DE3),诱导表达发现野生型BtDHX36蛋白(108 ku),结果显示目标蛋白在沉淀和上清液中均有表达,但主要存在于沉淀中(图2-C泳道1)。上清液样品经Ni-NTA亲和层析纯化可得到大量目的蛋白(图2-C泳道3),经sumo酶切后获得了不带sumo标签的蛋白(图2-C泳道4);经SP柱梯度洗脱后得到相对较纯的野生型BtDHX36(图2-C泳道5);最终浓缩得到纯度大于95%的野生型BtDHX36蛋白(图2-C泳道6)。

A.野生型BtDHX36菌落PCR鉴定结果,M.DS5000 DNA Marker,1.菌落PCR结果;B.野生型BtDHX36重组质粒的EcoRⅠ和XhoⅠ双酶切鉴定结果,M.DS5000 DNA Marker,1.双酶切产物;C.野生型BtDHX36蛋白诱导表达纯化结果,1.菌体破碎沉淀,2.菌体破碎上清液,3.Ni-NTA柱洗脱液,4.不带sumo标签的蛋白,5.Hi Trap SP HP离子柱纯化后的蛋白,6.浓缩结果,M.标准蛋白MarkerA.The wild-type BtDHX36 colony PCR identification results,M.DS5000 DNA Marker,1.Colony PCR results;B.Identification results of wild-type BtDHX36 recombinant plasmid by EcoRⅠ and XhoⅠ double digestion,M.DS5000 DNA Marker,1.Double digestion products;C.Induced expression and purification results of wild-type BtDHX36 protein,1.Bacterial crushing precipitate,2.Bacterial crushing supernatant,3.Ni-NTA column eluent,4.Protein without sumo tag,5.His Trap SP HP ion column purified protein,6.Concentration result,M.Standard protein Marker图2 野生型BtDHX36表达载体鉴定及蛋白的诱导表达和纯化Fig.2 Identification of wild-type BtDHX36 expression vector and induced expression and purification of protein
2.1.2 BtDHX36突变体表达载体的鉴定及蛋白纯化结果 BtDHX36突变体表达载体的鉴定及蛋白纯化结果见图3。

A.BtDHX36突变体菌落PCR鉴定结果,M.DS5000 DNA Marker;B.BtDHX36突变体重组质粒的EcoRⅠ和XhoⅠ双酶切鉴定,M.DS5000 DNA Marker;C.突变体浓缩结果,M为标准蛋白Marker;1~4.分别代表突变体BtDHX36R63AI65A、BtDHX36Y69A、BtDHX36K76AN77AK78A和BtDHX36Y862AA.PCR identification results of BtDHX36 mutant colonies,M is DS5000 DNA Marker;B.BtDHX36 mutant recombinant plasmid identified by EcoR Ⅰ and Xho Ⅰ double digestion results,M is DS5000 DNA Marker;C.Mutant concentration results,M is the standard protein Marker;1-4.Results of mutant BtDHX36R63AI65A,BtDHX36Y69A,BtDHX36K76AN77AK78A,BtDHX36Y862A,respectively图3 BtDHX36突变体表达载体鉴定及蛋白的诱导表达和纯化Fig.3 Identification of BtDHX36 mutant expression vector and induced expression and purification of protein
4种BtDHX36突变体表达载体经菌落PCR鉴定,均获得了与野生型BtDHX36一致的结果(图3-A)。挑选阳性菌落,提取质粒,经EcoRⅠ和XhoⅠ双酶切后,出现与野生型BtDHX36长度一样(2.8 kb)的片段(图3-B)。4种BtDHX36突变体经诱导表达纯化后,均获得与野生型一致的纯化结果,并得到纯度大于95%的BtDHX36突变体蛋白(图3-C)。
2.2 野生型BtDHX36解旋酶体外最佳底物结合条件的确定
由图4-A可知,当KCl浓度为50 mmol/L时,野生型BtDHX36解旋酶与16nt-ssDNA底物的平衡解离常数Kd值最小,为(16.2±0.1) nmol/L,表明此时蛋白与底物亲和力最强;由图4-B可以看出,当温度为37 ℃时,Kd值最小,因此BtDHX36结合底物的最佳反应温度为37 ℃;由图4-C可知,当MgCl2浓度为2 mmol/L时,Kd值最小,说明此时蛋白与底物亲和力最强;由图4-D可以看出,20 mmol/L Tris-HCl的pH过高或过低都不利于蛋白与底物的结合,当pH为7.5时,Kd值最小。综上可知,野生型BtDHX36与16nt-ssDNA底物的体外最佳结合条件为:KCl 50 mmol/L、MgCl22 mmol/L、20 mmol/L Tris-HCl pH=7.5、反应温度37 ℃。
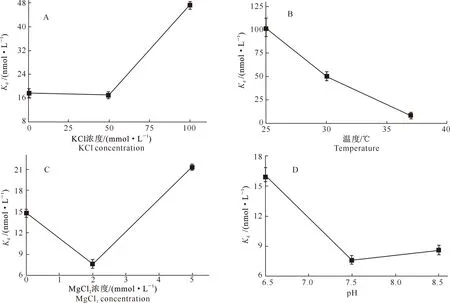
图4 野生型BtDHX36解旋酶与16nt-ssDNA底物的体外最佳结合条件Fig.4 In vitro optimal binding conditions of wild-type BtDHX36 helicase and 16nt-ssDNA substrate
2.3 野生型BtDHX36及其突变体与底物结合活性的比较
从图5-A可以看出,野生型BtDHX36蛋白、RSM区突变体蛋白(BtDHX36R63AI65A、BtDHX36Y69A、BtDHX36K76AN77AK78A)和OB域突变体蛋白(BtDHX36Y862A)均能高效结合Telomere G4底物。图5-B显示,野生型BtDHX36蛋白Kd值最小,为(56.5±0.4) nmol/L,表明其与底物的结合活性较突变体强;RSM区突变体蛋白BtDHX36R63AI65A、BtDHX36Y69A和BtDHX36K76AN77AK78A的Kd值较大,分别为(71.4±0.5),(75.8±0.2)和(62.7±0.4) nmol/L,表明其与底物的结合活性有所下降;OB域突变体BtDHX36Y862A结合该底物的Kd值最高,达到了(87.6±0.4) nmol/L,表明OB域位点Y862A突变的酶蛋白对底物结合活性有较明显影响。说明各突变位点均可降低酶蛋白与Telomere G4的结合活性。
2.4 野生型BtDHX36及其突变体解旋活性的比较
图6结果显示,野生型BtDHX36及其突变体对Telomere G4-16T均有解旋活性,但解旋比例存在差异,野生型蛋白、RSM区突变体BtDHX36K76AN77AK78A和OB域突变体BtDHX36Y862A的解旋比例接近80%,RSM区突变体BtDHX36R63AI65A和BtDHX36Y69A的解旋比例相对较低,分别为63%和51%。就解旋速率来讲,野生型BtDHX36、OB域突变体BtDHX36Y862A和RSM区突变体BtDHX36K76AN77AK78A的解旋速率常数均在0.40~0.50 s-1,差异不明显;RSM区突变体BtDHX36R63AI65A和BtDHX36Y69A的解旋速率在0.10~0.22 s-1,较野生型下降明显,表明这些位点的突变严重影响了BtDHX36蛋白的解旋速率。从以上结果可以看出,OB域突变对蛋白解旋影响不大,但RSM区不同位点的突变能不同程度地影响蛋白解旋的比例和速率,其中RSM区位点R63AI65A突变与Y69A突变的酶蛋白既影响了解旋比例又影响了解旋速率,表明这些位点在Telomere G4-16T底物解旋方面发挥着重要作用。

A.与底物结合比例;B.与底物结合的Kd值A.Binding fraction on Telomere G4 substrate;B.Kd value on substrate图5 野生型BtDHX36及其突变体与Telomere G4底物结合活性的比较Fig.5 Comparison of wild-type BtDHX36 and its mutants with Telomere G4 substrates binding activity

A.解旋动力学曲线;B.解旋速率常数A.Unbinding kinetic curves;B.Reaction rate constant图6 野生型BtDHX36及其突变体蛋白对Telomere G4-16T解旋活性的比较Fig.6 Comparison of the unbinding activity of wild-type BtDHX36 and its mutant proteins on Telomere G4-16T
3 讨 论
Chen等[27]研究发现,果蝇DHX36(DmDH-X36)的RSM区是特异性识别G4结构的重要部位。Srinivansan等[30]研究表明,RSM区不仅起到与G4结构亲和的作用,而且还能促进RNA双链和RNA-G4结构的重塑。BtDHX36与人类DHX36的序列同源性为91%,且研究表明,不同物种DHX36解旋酶的N端RSM区高度保守[31],这说明RSM区在不同物种间可能发挥相似的生物学功能,且这种功能是细胞生存所必需的。本试验比较了野生型BtDHX36与其突变体蛋白对Telomere G4-16T底物解旋活性的差异,发现相较于OB域氨基酸的突变,RSM区氨基酸突变对蛋白解旋Telomere G4-16T底物的影响更大,且RSM区位点R63AI65A突变与Y69A突变的酶蛋白均能在解旋比例和速率两方面影响解旋活性。这预示着在生物体内,BtDHX36解旋酶可能更倾向于在富含G4结构的端粒附近发挥生物学功能,且N端的RSM区在这种功能中起着无可替代的作用。
FA是一种根据荧光偏振原理研究生物大分子和核酸互作的一种手段,该方法具有灵敏性高、检测浓度低等优点[32-33]。本研究采用FA法,首先确定了BtDHX36与16nt-ssDNA结合的最适条件为KCl 50 mmol/L、MgCl22 mmol/L、20 mmol/L Tris-HCl pH=7.5、反应温度37 ℃;在此基础上检测了野生型BtDHX36及其突变体对Telomere G4底物的结合活性差异,结果发现,各突变体酶蛋白与Telomere G4的结合活性均低于野生型酶蛋白。FRET技术是近年发展起来的一项新技术,能在活细胞生理条件下对蛋白质-核酸间相互作用进行实时的动态研究。本研究利用FRET技术分析了野生型BtDHX36及其突变体对Telomere G4-16T解旋活性的差异,结果发现OB域Y862A突变对酶蛋白的解旋活性无明显影响,说明该位点可能不直接参与BtDHX36对含有G4结构底物的解旋活动;RSM区R63AI65A和Y69A突变均对酶蛋白的解旋活性有明显影响,降低了酶蛋白解旋含有G4结构底物的比例和速率,说明这2个位点是参与蛋白解旋G4结构的重要位点。
猜你喜欢
云南化工(2021年6期)2021-12-21 07:30:56
科学(2020年2期)2020-08-24 07:57:00
中国药理学通报(2019年10期)2019-09-24 08:07:46
蚕学通讯(2019年4期)2019-04-15 01:54:40
中国药理学通报(2018年7期)2018-07-04 11:15:46
物理学报(2018年11期)2018-06-19 10:04:22
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
生物技术通报(2015年1期)2015-04-10 16:15:19
山东医药(2015年40期)2015-02-28 14:28:45
