
WFS1基因突变致一个遗传性耳聋家系的临床表型及基因型分析
2021-05-25 02:51余奉徽王思霁代佳秋欧阳曦袁慧军耿佳卢宇康厚墉
中国听力语言康复科学杂志 2021年3期
余奉徽 王思霁 代佳秋 欧阳曦 袁慧军 耿佳 卢宇# 康厚墉#
作者单位:1 重庆医科大学附属第一医院耳鼻咽喉头颈外科 重庆 400010
2 陆军军医大学第一附属医院医学遗传中心 重庆 400038
3 四川大学华西医院罕见病研究院 成都 610041
耳聋作为世界上高发的、严重致残的疾病之一,给社会和家庭带来了经济和精神负担,其中约60%先天性耳聋与遗传因素相关。随着二代测序技术(next generation sequencing,NGS)在耳聋研究中的应用推广,耳聋基因的发现及变异检出发生了质的飞跃[1]。迄今为止,遗传性耳聋网站(http://hereditary hearingloss.org)已收录超过130个与耳聋相关的致病基因。其中WFS1基因在基因型及临床表型上具有高度异质性。目前已检测出超过200种发生于该基因的致病突变,虽然突变位点主要集中在8号外显子,但不同位点突变表现出不同临床表型,如Wolfram综合征、Wolfram样综合征、非综合征型感音神经性耳聋、单纯非胰岛素依赖型糖尿病等疾病。即使同一突变在不同人群、不同年龄段都可表现出不同的疾病特点[2]。上述复杂的基因型-表型相关性对于该基因的检测及诊断造成了一定困难和干扰。本文利用二代测序技术在一个遗传性耳聋家系中发现WFS1基因c.2051C>T(p.A684V)杂合突变,并对该位点的遗传特性、临床表型及族群分布特征等进行总结分析,以利于学者对该位点突变致病机理的探索及临床诊疗。
1 资料与方法
1.1 家系资料
该家系来自四川省,编号CQ10011,由重庆医科大学附属第一医院耳鼻喉科采集。采样小组对先证者及其亲属成员进行了问卷调查(包括现病史、既往史、母亲孕期情况及用药史、外伤史等)、听力学检查、体格检查及颞骨CT检查等。采集各家系成员外周血5 ml,提取基因组DNA[天根生化科技有限公司(北京)]于-80℃冻存备用。参与研究的家系成员均签署知情同意书,并通过重庆医科大学附属第一医院伦理委员会认可。
1.2 测序及变异位点致病性分析流程
使用定制的耳聋基因目标区域捕获试剂盒(Agilent)进行检测,该试剂盒覆盖目前已知耳聋基因158个。制备先证者DNA文库后,对158个耳聋相关基因全外显子和相邻内含子(±50 bp)区域进行捕获和富集,使用高通量测序平台(Illumina Hiseq X10)进行上机测序,以GRCh37/hg19为参考序列,测序下机数据应用BWA软件进行原始读长定位,Picard工具进行质控和去重操作,使用GATK和VEP工具进行变异识别和注释,最小等位基因频率(minor allele frequency,MAF)按0.5%进行变异过滤,结合先证者及其家系成员的临床表型和遗传方式筛查潜在的致病变异,并检索相关数据库(HGMD、Clinvar、DVD等),参考HL-EP2018版本ACMG遗传变异分类标准与指南对变异位点进行致病性判定。
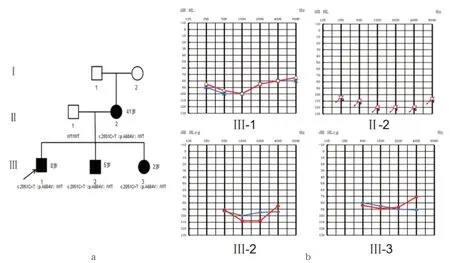
图1 a CQ10011家系图,黑色代表耳聋患者,箭头所示为先证者;b Ⅲ-1、Ⅱ-2为纯音听阈图,Ⅲ-2 、Ⅲ-3为ASSR反应阈图;红色代表右耳,蓝色代表左耳。

图2 Sanger测序图谱
1.3 Sanger测序验证
针对检出的WFS1基因突变位点设计验证引物,对已采集样本的家系成员进行Sanger测序验证。测序数据通过Mutation-Surveyor软件进行序列比对,分析检出变异是否符合家系内听力表型和基因型共分离。
2 结果
2.1 家系资料及听力学特征
CQ10011家系为彝族,共追溯回访3代、7人,除Ⅰ-1、Ⅰ-2外,其余人员均行基因检测,其中耳聋患者4人、均表现为先天性耳聋。根据世界卫生组织1997年耳聋分级标准,先证者Ⅲ-1(8岁)及Ⅱ-2(41岁)纯音听阈检测结果提示双耳极重度感音神经性聋,Ⅲ-2(5岁)、Ⅲ-3(2岁)患者ABR及ASSR检测结果提示双耳极重度感音神经性聋。Ⅲ-1、Ⅲ-2、Ⅲ-3行颞骨CT检查未见明显结构异常。上述耳聋患者否认耳鸣、眩晕、视物模糊、繁渴、多饮、多尿及体重下降等不适,否认外伤、耳毒性药物及噪音接触史,否认近亲婚育史及母亲孕期特殊疾病史;耳科、眼科专科查体及系统体格检查未见明显外观异常(图1)。
2.2 高通量测序及结果分析
对先证者进行靶向捕获高通量测序,平均测序深度126×,10×以上覆盖度96%。下机数据通过质控和去重后,使用GATK和VEP进行变异识别和注释。以MAF值<0.5%进行变异位点过滤,根据听力表型、VEP Impact注释得分等条件进行筛选,在已知耳聋基因中检出WFS1基因NM_006005.3:c.2051C>T(p.A684V)杂合突变,为已报道的致病突变。
2.3 sanger测序验证及基因型-表型共分离分析
先证者Ⅲ-1及其弟弟Ⅲ-2、妹妹Ⅲ-3均携带WFS1基因c.2051C>T(p.A684V)杂合突变;先证者父亲为野生型,母亲携带c.2051C>T杂合突变。先证者Ⅲ-1及其弟弟Ⅲ-2、妹妹Ⅲ-3的基因突变来源于母亲,符合家系内表型和基因型共分离,符合常染色体显性遗传(图1、图2)。
3 讨论
本研究中,笔者检出WFS1基因c.2051C>T(p.A684V)杂合突变,该突变在正常人群中罕见,为目前已报道的耳聋致病基因,先证者临床表型与WFS1基因致病的临床表型相符合,家系内表型和基因型共分离。根据美国医学遗传学与基因组学学会(the American college of medical genetics and genomics,ACMG)遗传变异分类标准与指南分析,明确该突变为该家系致病突变。
WFS1基因位于染色体4p16.1,由8个外显子组成,基因组DNA跨度为33.4 kb,其中1号外显子不编码。该基因编码的wolframin蛋白定位于内质网,在细胞膜运输、分泌、加工或调节内质网Ca2+稳态方面具有重要生理功能[3]。WFS1基因在基因型及临床表型上具有高度异质性。目前已知WFS1基因突变可致非综合征型显性遗传耳聋,主要以迟发性低频下降为主;还可致非综合征型隐性遗传耳聋,主要以先天性极重度感音神经性聋为主,另外可致Wolfram综合征、Wolfram样综合征、单纯非胰岛素依赖型糖尿病等疾病。Heredia等[2,4]回顾性分析了1998~2013年发表的由WFS1基因突变致Wolfram综合征患者的临床表型及遗传数据,总结该综合征的耳聋主要表现为语后渐进性感音神经性聋,发病年龄中位数为12.5岁(5~39岁);其他主要症状还包括糖尿病、视神经萎缩、尿崩症及神经、精神疾病等。
2001年意大利Tessa等[5]首次报道WFS1基因c.2051C>T(p.A684V)突变致儿童期Wolfram综合征,随后不同地区的多位学者相继报道该位点突变致非综合征型耳聋等不同临床表型[6~11]。笔者结合本研究病例对该位点已报道的文献进行分析总结,其主要致病特点总结见表1。
表1可知,除20号病例表现出Wolfram综合征典型症状外,其余患者均以感音神经性聋为首发症状,且发病年龄均小于3岁,表现为语前聋、全频受损、受损程度为重度至极重度,需要及时的听力检查及干预措施。第2个出现的症状为视神经萎缩,发病年龄中位数为12岁(3~41岁),这与典型的Wolfram综合征患者视神经萎缩的发病年龄基本吻合[4]。另有少部分病例表现出尿崩症及精神神经系统症状;除20号病例外,均未发现糖尿病相关症状。
同时笔者观察到一个有趣的现象,在现有病例中,中国、日本等东亚地区的病例仅表现为非综合征型感音神经性聋,未观察到糖尿病、视神经萎缩及尿崩症等相关病变,而欧美地区病例以Wolfram样综合征为主。这提示笔者可能不同人群遗传背景或地理环境因素对该致病位点的致病性有重要影响因素,这对该基因致病机理的阐明有重要启示作用。在本家系及其余中国和日本病例中,除1号及9号病例外,其余病例报道时年龄均较小、未达到视神经萎缩发病年龄中位数,是否出现视神经萎缩及糖尿病等其它症状仍需继续观察,这也提示对于该类病例进行视神经萎缩的相关眼科检查和糖尿病的相关内分泌科检查及随访的必要性。
Guan等[10]对中国128个非血缘关系的非综合征型感音神经性耳聋散发性病例进行基因筛查,发现5例WFS1基因突变致耳聋,其中c.2051C>T (p.A684V)4例、c.2590G>A (p.E864K)1例,从而推测c.2051C>T可能为中国人群的热点突变。本研究与该突变位点及临床表型吻合,进一步支持该推测的可能性。同时也观察到在这20例病例中至少有8例为新发突变,从而推测在本研究中先证者母亲可能也是新发突变,因此其父母都没有耳聋表型。

表1 20 例WFS1 基因c.2015c>T 突变病例特征总结
综上所述,笔者通过二代测序技术鉴定出WFS1基因c.2051C>T(p.A684V)杂合突变为该家系耳聋致病基因,且该结果进一步验证了c.2051C>T突变可能为WFS1基因在中国人群中的热点突变,该致病突变在中国人群中主要临床表型为语前非综合征型感音神经性聋。不排除将来表现为综合征的可能性,应当注意随访其他系统的表型。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国现代医生(2022年21期)2022-08-22
农村科学实验(2022年2期)2022-03-12
医学前沿(2021年18期)2021-04-14
三农资讯半月报(2020年2期)2020-03-09
家庭百事通·健康一点通(2019年8期)2019-08-29
森林工程(2018年1期)2018-05-14
农村百事通(2017年3期)2017-03-10
