
基于多数据源与机器学习的药物副作用预测
2021-05-25 05:26杜瑶
软件导刊 2021年5期
杜 瑶
(上海理工大学管理学院,上海 200093)
0 引言
药物不良反应(Adverse Drug Reaction,ADR)通常定义为在正常药物剂量下,在预防、诊断或治疗中产生的有害和非预期的影响[1]。这种影响会给患者身心造成危害,严重的甚至会威胁到患者生命安全。近年来,因药物副作用所引发的药物安全问题备受关注。在美国,每年有200 万人受到药物副作用的影响,其中有10 万人因严重的药物副作用而死亡[2]。严重的药物副作用也是新药开发失败和已上市药物撤回的一个重要因素[3]。由于药物研发周期过长以及药物研发所需的人力物力和经济成本非常高昂,所以提前预测药物副作用可起到指导药物开发的重要作用,针对已上市药物的副作用进行预测还可有效指导医生进行临床治疗。因此,及时、准确地预测药物副作用已成为国内外研究的热点问题[4]。
近年来,众多研究人员也提出了一些预测药物不良反应的方法。例如,Liang 等[5]在2019 年利用多视图学习重要特征进行药物副作用预测;Hu 等[6]在2018 年利用堆叠深度异构信息网络嵌入方法进行药物副作用预测;Emir等[7]在2017 年将生物医学知识以图的形式生成不同特征集以预测药物副作用;Zheng 等[8]在2018 年利用药物特征的逆相似性构建可靠的负样本以预测药物副作用。以上都是使用计算方法对药物副作用进行预测。因此,利用数据挖掘和机器学习等计算方法分析海量的生物医学数据,实现对药物副作用的精准预测已成为了该领域的热门研究方法[9]。很多将药物自身特征作为研究的主要对象,这些特征包括药物的化学结构、靶点蛋白、分子通路等。综上所述,整合药物副作用相关信息,利用当下流行且高效的计算方法开发一套药物副作用预测工具,既可以帮助药物研发人员进行新药研发,也可以帮助患者规避临床治疗中产生副作用的风险,具有重要的科学意义及广阔的应用前景[4]。
1 预测方法
1.1 原理
通常认为在药物的作用下,如果基因表达量与患病情况下的基因表达量变化情况相反,则认为药物起到了积极作用,如果基因表达量与患病情况下的基因表达量变化情况相同,则认为药物可能没有起到积极作用,或者说药物更可能产生副作用,因为其导致了基因的非正常表达,这也是本文的核心思想。此前,也有不少学者使用基因相关信息作为研究对象建立预测模型。例如,Xiang 等[1]利用基因与药物副作用的关联网络进行药物副作用预测;Uner等[10]将基因表达作为特征,使用深度学习框架进行药物副作用预测;Fukuzaki 等[11]利用统计相似性模型,基于药物通路和基因表达谱信息预测药物副作用;Wang 等[12]在2016 年利用LINCS L1000 经典基因表达数据集结合化学结构等信息进行药物副作用预测。但以上预测方法都未将基因表达量变化情况与疾病联系起来。在疾病治疗过程中,用药后即会引起体内基因的表达量发生变化,一旦出现副作用,体内某些基因就会出现非正常表达,也即是说副作用与基因表达始终是联系在一起的。因此,基于基因表达的副作用预测方法就是将药物副作用与基因表达联系起来,找到两者之间的关联性。本文利用患病前后和用药前后重要基因的表达量变化情况作为解释变量,因变量副作用标签来自权威的SIDER 数据库,再利用当下流行且高效的机器学习算法建立模型进行药物副作用预测。
1.2 工作内容
本文首要进行的工作是准备好高质量的数据,本文数据全部来自官方数据库。从官方数据库中下载所需的原始数据,包括患病基因表达数据集、用药基因表达数据集、人类重要基因集以及药物副作用数据集。本文以乳腺癌细胞作为研究对象,因此从数据集中选择与乳腺癌相关数据,在数据集预处理过程中,按照需要对原始数据进行合理筛选。经过层层筛选后得到所需数据,最终选定了401种药物和142 个基因用于模型构建,标签则是一个表示副作用有无的401 维向量。
在完成数据预处理后,还要筛选适合本文数据类型的机器学习算法建立模型。这里根据数据类型及数据量大小选择了随机森林算法(Random Frost,RF),并且选择K 近邻(k-Nearest Neighbor,KNN)、决策树(Decision Tree)以及朴素贝叶斯(Naive Bayesian,NB)3 个算法作为对比。算法确定之后,再利用处理好的特征数据进行模型训练与测试,然后对比每种机器学习模型所得结果,最终发现随机森林在几种算法中表现最好。
2 特征工程
2.1 数据收集及预处理
GEO 和cmap 两个基因表达数据库是本文的核心数据库,其中基因受到疾病影响所产生的基因表达量变化情况来自GEO 数据库(https://www.ncbi.nlm.nih.gov/),这是美国国立卫生研究院1988 年创立的美国国立生物技术信息中心,目的是给分子生物学家提供一个信息存储与处理的系统。在数据库中找到所需的样本组,这里选择的是12 个乳腺细胞样本,分为6 个正常对照组和6 个患病实验组。Value 值即为样本某个探针所对应基因的表达量,如表1所示。基因的差异表达分析是利用R 语言的Limma 包实现的,这是一个专门用于基因差异分析的包[13]。
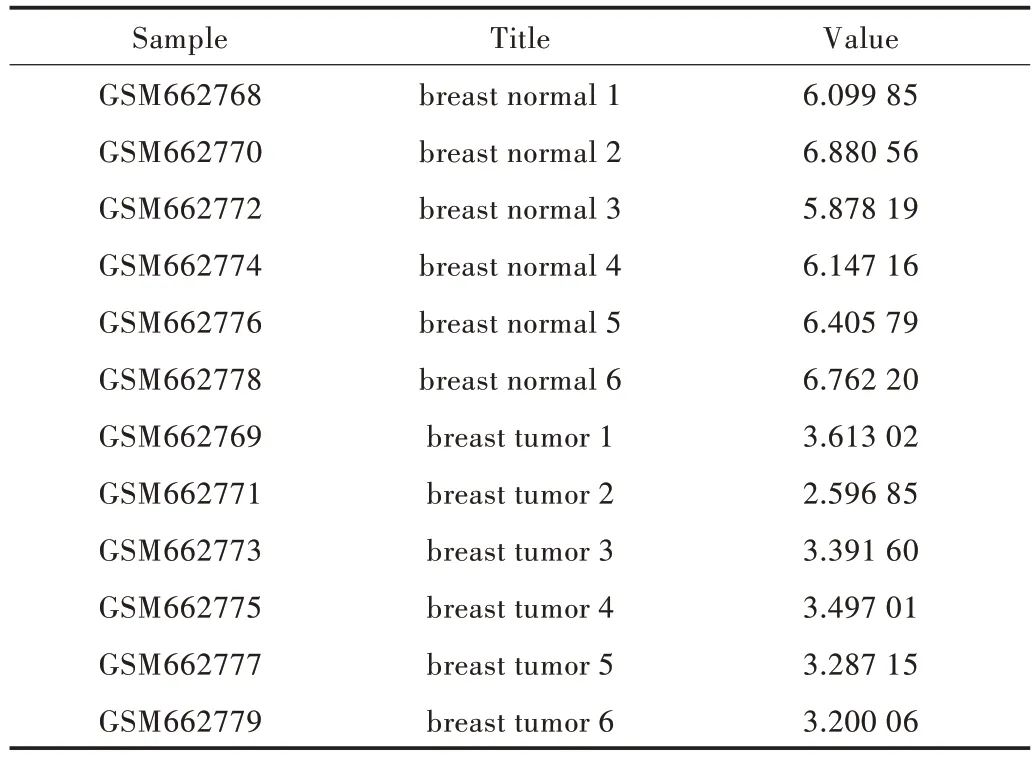
Table 1 Breast cell sample data表1 乳腺细胞样本数据
本文选择直接在数据库官网在线对两组样本的基因表达量差异进行分析,由于这里的差异分析同样是使用R语言的Limma 包得出的结果,所以该结果真实可靠,一共得到2 万多个基因的差异表达。以下呈现了前5 个基因的差异分析结果,如表2 所示。

Table 2 Results of difference analysis表2 差异分析结果
但这里得到的差异分析结果并不能直接使用,还要对其进行筛选。在统计学上,adj.P.Val 的值越小,表示结果越好。通常情况下,本文设置adj.P.Val 的阈值为0.05,取小于等于0.05 的值[14]。logFC 表示差异量,绝对值越大,表示两组样本的基因表达量差异越大,大于零表示后者的表达量大于前者,小于零表示后者的表达量小于前者。其值由如下公式求出:

式中,x为实验组样本的基因表达量,y 为对照组的基因表达量。这里log(FC)的阈值设置为1,筛选出实验组基因表达量为对照组基因表达量至少两倍的基因作为差异表达基因。log(FC)大于零的部分基因作为在疾病作用下上调的基因,小于零的部分基因作为在疾病作用下下调的基因。在对数据进行筛选的过程中,还需要用到一个对重要基因进行筛选的数据库(http://tubic.tju.edu.cn/deg/blast.php?type=single&db=e)。这些基因的表达量变化会对人体产生较大影响,因此本文重点关注这些基因的表达量变化情况。将重要基因数据集与之前预处理的基因集合取交集,便得到患病之后重要基因的表达量变化情况。
接下来是对用药之后可体现基因表达量变化的相关数据进行处理,这里选择的是cmap 数据库(https://portals.broadinstitute.org/cmap/)中药物对疾病细胞的处理结果。cmap 数据库是一个基因表达谱数据库,利用药物小分子对疾病细胞进行处理,得到一个前后基因表达量变化谱。为了与前面的疾病相对应,同样选择乳腺癌细胞的处理结果。本文将ratio 矩阵中作用于乳腺癌细胞的部分筛选出来,进行相应处理后,形成一个药物与基因表达量的矩阵,每个药物与每个基因作一次映射。ratio 矩阵中大于1 的值表示用药后基因表达量上调,反之下调。最后同样将基因集与此前处理的疾病情况下上下调基因结果取交集,得到最终的基因集合。
2.2 特征构建
在最终数据集的处理中,本文将患病与用药之后同时上调的基因标记为1,同理,将患病与用药之后同时下调的基因也标记为1,反向变化的则标记为0。最终的解释变量形式如表3 所示。一个药物样本的形式为一串0、1 数字的组合,表示在药物作用下特定基因的表达量变化情况。这里考虑同时上调和下调都属于患病与用药之后的同向变化,因此把同时上调和下调的基因组合到一个矩阵里,将药物作用下同向变化的不管是同时上调还是同时下调的基因都标记为1,而反向变化的不管是先下调再上调,还是先上调再下调,都标记为0。假设此为矩阵T,如果=1,表示基因j在疾病和药物i的作用下,表达量发生了同向变化。
因变量也即本文的数据标签,来自权威的药物及相关副作用资源库SIDER 数据库,里面收集整理了1 430 个药物、5 880 个副作用信息以及99 423 个药物—副作用关系[4]。这里将此1 430 种药物与之前cmap 数据库中作用于乳腺癌细胞的1 241 种药物取交集,得到401 种药物集合。这401 种药物便是解释变量的样本数量,标签则是这401 种药物所对应的有无某副作用的情况,数据形式是一个401 维的向量,也用0 和1 表示,1 表示药物有该副作用,0 则表示药物没有该副作用。

Table 3 Results of data processing表3 数据处理结果
3 分类器选择
3.1 选择依据
构建好模型特征之后,选择一种合适且高效的算法也是本文工作的重要内容,目前最流行的是使用机器学习算法建立模型进行分类预测等任务。如今,机器学习、深度学习等技术已得到了广泛应用,相关算法也因不断优化使得代码更加通俗易懂、易于实现,可根据数据类型、数据量大小以及任务内容进行算法选择。
本文数据特征是一个药物样本的142 个基因在患病前后和用药前后的基因表达量变化情况,其是由0 和1 的布尔类型数据组成的,并非具体某个数值。通过分析,可判断这种数据类型不适合类似线性回归的相关算法。由于复杂的算法往往需要大量数据才能发挥效果,本文数据量不多,而且是一个二分类问题,因此需要尽量选择简单、高效的分类算法。本文选择的目标分类器是随机森林,其是一种集成学习算法,可整合多棵决策树,从中得到最优结果,另外还选择了决策树算法与随机森林算法进行对比,验证随机森林融合多棵决策树取最优结果的特点,最后使用具有概率特点的朴素贝叶斯作为本文的分类器。最终,本文选择了随机森林、K 近邻、决策树、朴素贝叶斯4种机器学习算法。
3.2 分类器介绍
3.2.1 随机森林
随机森林算法(Random Frost)是基于Bagging 集成学习理论的代表算法,由Breiman[15]于2001 年提出。其是利用bootsrap 重抽样方法从原始样本中抽取多个样本,对每个bootsrap 样本进行决策树建模,然后组合多棵决策树的预测,通过投票得出最终预测结果。一棵决策树的分类能力可能很弱,但在随机产生大量决策树并组成随机森林后,每个样品都逐一通过一棵树分类决策,最后组合的结果将更接近于正确分类[16]。大量理论与实证研究都证明了RF 具有很高的预测准确率,对异常值和噪声具有很好的容忍度,且不容易出现过拟合。随机森林通过在每个节点处随机选择特征进行分支,每棵分类树之间的相关性得到最小化,故对多元共线性不敏感,从而提高了分类精确性及抗噪声能力[17]。随机森林是一种自然的非线性建模工具,也是目前数据挖掘领域的热门研究方向之一。
3.2.2 K 近邻
K 近邻算法(KNN)是基于实例的代表算法,于1967 年由Cover 等[18]提出,是一种用于分类与回归的统计方法,主要思想是通过测量不同特征值之间的距离进行分类。如果一个样本在特征空间中的k 个最相似或在特征空间中最邻近样本中的大多数属于某一类别,则该样本也属于该类别。在KNN 算法中,所选择的邻居都是已正确分类的对象。该方法在确定样本类别时依据最邻近的一个或几个样本类别决定待分样本所属类别[17]。在本文工作中,观察离它最近的一个或几个药物的基因表达特征标签属于哪一类,则目标样本也属于此类。
3.2.3 决策树
决策树,顾名思义,其类似于一棵树,利用树的结构对数据记录进行分类,树的一个叶结点即代表某个条件下的一个记录集,根据记录字段的不同取值建立树的分支,在每个分支子集中重复建立下层结点和分支,便可生成一棵决策树[19]。本文的数据特征为离散变量,非常适合使用决策树对数据作分类预测,同理也非常适用于随机森林算法。
3.2.4 朴素贝叶斯
1960 年,Maron 等[20]首先提出朴素贝叶斯分类方法,其是一种基于概率模型的分类方法。概率模型即为贝叶斯概率公式,即:
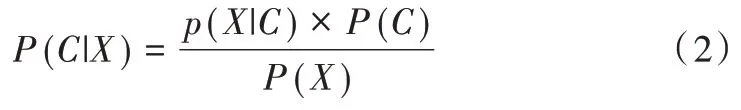
其中,P(C|X)为条件X 下C 的后验概率,P(C)为C的先验概率,P(X|C)为条件C 下X 的后验概率,P(X)表示X 的先验概率[21]。本文中C 为类别变量,X 为样本变量,则贝叶斯概率公式就是求解样本属于某一类别的概率,然后选择概率值最大的类别作为模型分类结果。
3.3 模型实现
本文模型是基于目前流行的Python 机器学习库sklearn 实现的。由于样本数量少,因此本文随机选择90%的样本作为训练集,剩下的10% 作为测试集。在机器学习任务中,需要尽量保证正负样本的均衡,才能使模型具有较好的泛化性能。因此,从整理好的SIDER 数据库中选择正负样本相对均衡的5 种副作用应用于本文模型,得到的测试集准确率结果如图1 所示(彩图扫OSID 码可见)。

Fig.1 Results of model prediction图1 模型预测结果
从图1 中可以很直观地看出,在副作用预测任务中,随机森林算法得到的准确率明显优于K 近邻算法,并且在5 种副作用测试结果中都是最优的,这也说明本文数据在随机森林中的表现相比其它3 种算法更好。在副作用皮疹的测试结果中,随机森林的准确率达到90.24%,而K 近邻只有58.54%。在副作用口腔炎的测试结果中,随机森林的准确率为78.05%,但也依然优于其它3 种算法。这一结果验证了本文的研究思路,证明了随机森林分类器优越的分类能力,也充分说明基因表达量变化情况与药物副作用之间具有很强的相关性。
4 结语
本文以患病前后与用药前后的基因表达量变化情况为研究对象,分析了基因表达量变化情况与药物副作用的关联关系,提出一种药物副作用预测方法。由于数据集中某些数据的缺失,以及对数据进行的层层筛选都导致了本文数据集的大幅减少,这对模型的构建会产生影响,因此数据集的扩展是本文需要突破的一个方面。由于生物信息数据存在重复样本的特点,若在数据预处理过程中遇到重复样本时采用平均处理方法,以及两次基因表达量测试是在不同平台上进行的,这都会导致数据出现误差。另外,本文只使用了基因表达这一单一特征以及简单的机器学习算法,因此对特征的融合以及算法选择也是本文下一步要突破的方向。
猜你喜欢
肝博士(2022年3期)2022-06-30
数学小灵通(1-2年级)(2021年4期)2021-06-09
成都信息工程大学学报(2019年3期)2019-09-25
中学生数理化·七年级数学人教版(2019年4期)2019-05-20
电子制作(2018年16期)2018-09-26
中学生数理化·七年级数学人教版(2018年6期)2018-06-26
初中生世界·七年级(2017年9期)2017-10-13
中央民族大学学报(自然科学版)(2016年4期)2016-06-27
特别健康·上半月(2016年2期)2016-05-30
郑州大学学报(医学版)(2015年1期)2015-02-27
