
基于TCGA数据库筛选调控mRNA表达的子宫颈癌相关基因
2021-05-18 01:35石凤陈志鸿李光景陈升才罗小琼王俊利
中国医科大学学报 2021年4期
石凤,陈志鸿,李光景,陈升才,罗小琼,王俊利
(右江民族医学院 1.附属医院生殖医学中心;2.基础医学院,广西 百色 533000)
子宫颈癌是女性最常见的恶性肿瘤之一,世界卫生组织报道子宫颈癌的发病率和死亡率有下降趋势,但每年仍有750 000例新增患者及311 000例患者死于子宫颈癌[1]。子宫颈癌致病因素涉及高危型HPV感染、表观遗传改变和基因序列突变。研究[2]表明多种生物过程的基因变异参与了子宫颈癌的发生发展。基因遗传变异使患者对疾病的易感性和患病后的严重程度各不相同,从分子生物学水平研究子宫颈癌诊断和预后对于患者的个体化和精确治疗至关重要。
基于高通量测序的生物信息学研究癌症的差异表达基因、初步筛选与癌症相关的早期分子诊断和治疗靶点是目前重要的手段之一。单核苷酸多态性(single nucleotide polymorphism,SNP)是多种癌症的治疗靶点,对于癌症的早期诊断和治疗具有重要意义,一些基因的SNP被矫正可能会逆转癌症的恶性生物学行为。随着全基因组序列的研究进展,越来越多与子宫颈癌易感性相关的SNP被发现[3-5]。癌症基因组图谱(the cancer genome atlas,TCGA)数据库广泛应用于癌症相关基因的高通量基因组分析,初步筛选与癌症相关的基因。本研究通过对TCGA数据库中与子宫颈癌相关的基因SNP数据进行生物信息学分析,筛选与子宫颈癌相关的突变基因,旨在为阐明子宫颈癌发病机制提供参考。
1 材料与方法
1.1 子宫颈癌SNP数据来源
TCGA数据库SNP原始数据不对外开放,因此从TCGA数据库(https:// portal.gdc.cancer.gov/)下载经处理后的子宫颈癌SNP相关数据,同时下载子宫颈癌mRNA原始数据,共309个mRNA样本,包括3个正常样本和306个肿瘤样本。从SNP相关数据得到子宫颈癌突变基因,利用R语言中的Edger包对mRNA数据进行整合和标准化,得到差异表达基因以及表达水平。
1.2 差异表达SNP功能富集分析
通过DAVID软件对子宫颈癌样本突变>20例的突变基因进行基因本体论(gene ontology,GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析,以P< 0.05为检验水准,获得突变基因的分子功能(molecular function,MF)、生物过程(biological process,BP)、细胞组分(cellular component,CC)和KEGG通路信息。
1.3 差异表达SNP的mRNA表达
对mRNA整理和标准化来获得mRNA表达水平,对突变例数多的基因进行分析,通过秩和检验得到基因突变与基因mRNA表达水平的相关性,对具有相关性的基因进行生存曲线分析并绘制生存曲线。
1.4 统计学分析
子宫颈癌差异基因使用R软件3.6.0“Edger”包进行筛选,通过Wilcox秩和检验比较突变基因mRNA的表达水平,通过Kaplan-Meier绘制生存曲线。P< 0.05为差异有统计学意义。
2 结果
2.1 子宫颈癌SNP数据分析
结果显示,有110个基因突变样本超过20例(图1)。共309个子宫颈癌mRNA样本,癌症样本有306个,设定阈值为4,P< 0.01,对正常组织和子宫颈癌组织样本进行差异表达分析,共获得803个差异表达基因(图2)。

图1 突变>15例的突变基因瀑布图Fig.1 Waterfall map of mutated genes from more than 15 samples
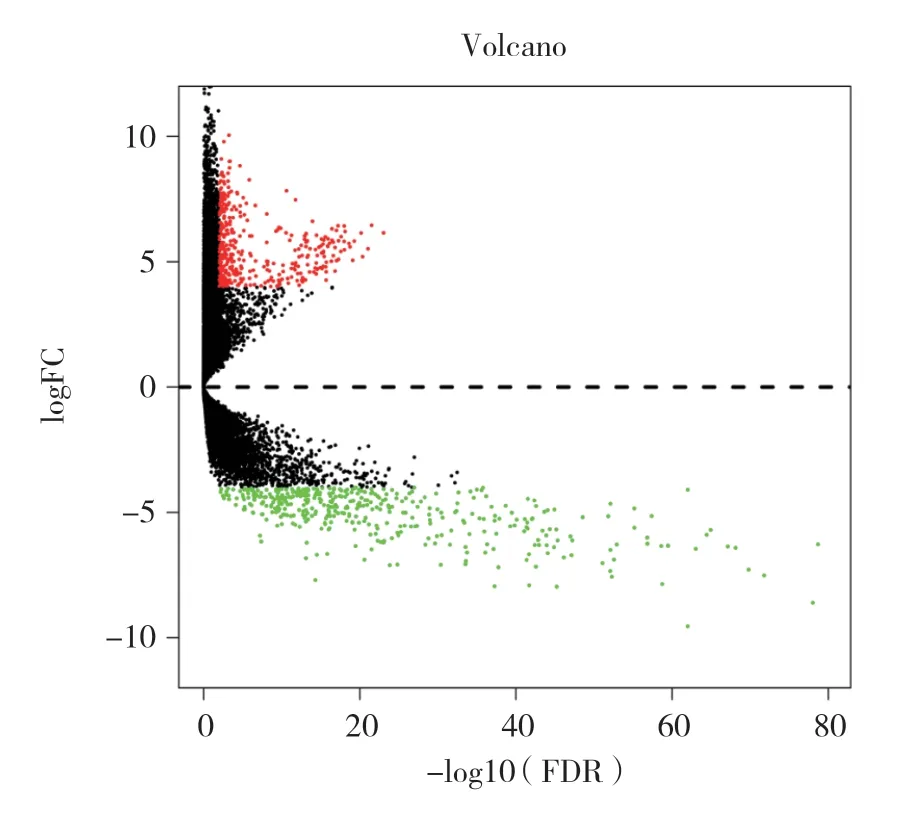
图2 差异表达mRNA的火山图Fig.2 Volcanic map of differentially expressed mRNA
2.2 GO和KEGG富集分析
结果显示,子宫颈癌突变基因参与多种通路途径影响BP和MF。在BP方面,SNP主要影响转录过程,参与RNA聚合酶Ⅱ的负调节和细胞膜黏附分子的黏附能力;在CC中,SNP主要定位于细胞膜和细胞外泌体。在MF方面,SNP主要参与蛋白质结合、Ca2+离子结合、ATP酶结合和激活,见表1。此外,对突变基因进行KEGG富集分析结果显示,SNP参与多条信号通路(甲状腺激素信号通路和Notch信号通路等),见表2。
2.3 突变基因蛋白互作网络(protein-protein interaction networks,PPI)构建及其与mRNA表达水平的相关分析
为了了解蛋白质互作关系,通过String在线软件对110个突变基因构建PPI,包括107个节点和299个边。通过Cytoscape软件的cytoHubba包进行可视化处理,见图3。对突变基因和经校正后的mRNA表达进行相关性分析。结果显示,DNAH17、FBXW7和SYNE23个与mRNA表达相关的突变基因,DNAH17和SYNE2突变后的mRNA表达水平降低,而FBXW7突变后mRNA表达水平升高(P分别为0.020、0.029、0.031),见图4。对DNAH17、FBXW7和SYNE2突变基因绘制Kaplan-Meier生存曲线,发现SYNE2高表达显著降低子宫颈癌患者的无病生存期(P=0.008),见图5。

表1 子宫颈癌突变基因的功能富集分析Tab.1 Functional enrichment analysis of cervical cancer gene mutations

表2 子宫颈癌突变基因KEGG信号通路富集分析Tab.2 KEGG signaling pathway analysis of cervical cancer gene mutations
3 讨论
SNP指单碱基DNA序列发生突变、逆转、插入和缺失,是人类最常见的遗传变异方式,基因启动子SNP变异可影响RNA聚合酶识别转录起始位置,使mRNA表达水平异常,基因内含子区域SNP影响mRNA可变剪切过程[6]。研究表明,基因突变与子宫颈癌的发病风险有关。DUAN等[7]发现IL-6 -174G>C降低子宫颈癌的发病风险。HABBOUS等[8]发现P53Arg72Pro突变促进HPV阳性患者的子宫颈病变,增加子宫颈癌患病风险。

图3 子宫颈癌突变基因的PPI网络Fig.3 PPI networks of cervical cancer mutant genes

图4 子宫颈癌突变基因与mRNA表达水平的相关性Fig.4 Correlation between cervical cancer gene mutations and mRNA expression levels

图5 子宫颈癌突变基因的无病生存期(Kaplan-Meier)Fig.5 Disease-free survival in patients with cervical cancer gene mutations(Kaplan-Meier)
本研究从TCGA公共数据库下载子宫颈癌相关SNP数据和表达谱数据,通过表达谱数据获取经校正后的基因表达水平和差异表达基因,通过SNP数据筛选出突变基因。为了阐明这些突变基因在疾病中的分子机制,本研究通过GO和KEGG富集分析结果显示,这些突变基因主要定位于细胞膜和细胞外泌体,参与蛋白质结合和ATP酶激活,并参与转录调控作用,包括负调节RNA聚合酶Ⅱ启动子转录。此外,通路分析表明这些突变基因参与Notch信号通路、甲状腺激素信号通路、细胞周期、病毒致癌和多种癌症致病过程。
对突变>20例的突变基因表达水平分析结果显示,DNAH17和SYNE2突变后的mRNA表达水平降低,而FBXW7突变后的mRNA表达水平升高。进一步对DNAH17、SYNE2、FBXW7基因进行生存分析发现,SYNE2高表达显著降低子宫颈癌患者的无病生存期(P< 0.05),然而,未发现DNAH17和FBXW7基因与子宫颈癌生存期有关,可能是在TCGA数据库子宫颈癌相关SNP数据中,基因突变例数过少,导致生存时间差异不显著。DNAH17是与轴突重链编码的相关基因,FAN 等[9]发现DNAH17的异常甲基化水平与肝癌纤维胶囊、肿瘤坏死、肝硬化和肿瘤血栓等临床特征有关。此外,ZHAN等[10]对乙型肝炎病毒相关的早期肝癌组织进行整个外显子进行了测序,发现DNAH17在肝癌中存在高频突变,与本研究通路富集结果一致。子宫颈癌的发生发展主要是HPV持续感染致癌的动态过程,本通路富集分析表明突变基因参与病毒致癌。目前,尚未发现突变基因与子宫颈癌相关性的研究报道,本研究为子宫颈癌的分子机制研究提供了新的理论依据。
FBXW7是包含F框和WD重复域蛋白,属于F盒蛋白家族,是SCFE3泛素连接酶底物识别部位[11]。FBXW7参与细胞调控(细胞周期、细胞分化、细胞增殖和凋亡)。BEN等[12]发现miR-27a-3p通过下调FBXW7促进子宫颈癌细胞增殖。XU等[13]研究发现FBXW7表达水平降低与淋巴血管间隙浸润和淋巴结转移有关,提示患者无病生存期和总体生存期较短。本研究通路富集表明突变基因参与子宫颈癌致癌过程,FBXW7突变使mRNA表达水平升高。因此,突变型FBXW7可能降低子宫颈癌的恶行生物学行为。SYNE2属于巨谱蛋白重复序列(Nesprins)家族,主要参与连接细胞核与细胞骨架。研究[14]表明SYNE2突变rs4027405与抑癌基因P21表达有关,携带rs4027405 GA/AA基因型的肝癌患者生存期较短、预后较差。本研究中发现SYNE2突变后表达水平降低,且低表达SYNE2的子宫颈癌患者无病生存期较长。
综上所述,通过GO和KEGG富集分析发现DNAH17、FBXW7和SYNE2参与多种致癌过程。子宫颈癌组织中DNAH17、FBXW7和SYNE2突变调控mRNA的表达水平,且SYNE2突变患者无病生存期较长,表明SYNE2突变是子宫颈癌的保护因素。本研究为临床诊断和预后评估提供了新的思路,但仍需在今后的临床研究中进一步验证。
猜你喜欢
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
昆明医科大学学报(2021年1期)2021-02-07
天津医科大学学报(2021年1期)2021-01-26
康颐(2020年13期)2020-11-10
现代养生·上半月(2020年4期)2020-04-20
中国保健营养(2019年4期)2019-09-10
中国现代医生(2016年31期)2017-03-02
中国现代医生(2016年32期)2017-03-02
医学研究杂志(2015年12期)2015-06-10
