
全固态锂金属电池多物理场耦合下的电化学过程仿真模拟
2021-05-17 08:55:40孙哲韬何英杰陈邵杰黄缘齐
高等学校化学学报 2021年5期
孙哲韬,何英杰,陈邵杰,聂 璐,黄缘齐,刘 巍
(上海科技大学物质科学与技术学院,上海201210)
快离子导体,又称固体电解质,作为一种固态材料其拥有接近甚至高于液体的离子电导[1]. 使得快离子导体能够在许多高新科技领域得到应用,如固态电池、燃料电池、忆阻器及气体传感器等[2~4]. 而室温下离子电导率大于10 mS/cm的快锂离子导体的发现使得比液态锂离子电池具有更高理论比容以及更快充电速率的固态锂离子受到关注. 固态电解质包含多种类型. 如由无机晶态物质组成的石榴石型、锂快离子导体型、钠快离子导体型与钙钛矿型固体电解质等;由非晶态硫化物组成的Li2S为主体的多元非晶固体电解质,氮氧化锂磷(LiPON)等非晶固体电解质以及聚合物固体电解质等[5].
锂金属作为电化学势最低[-3.05 V(vs. SHE)]的负极,具有极大的理论能量密度(3860 mA·h·g-1),是传统石墨负极的10倍[6]. 作为下一代高能锂电池的理想材料,在锂金属负极参与实际生产应用前需克服一些安全性与稳定性的挑战[7]. 主要归因于在充放电循环中,锂离子并不会均匀沉积在负极表面,而这种体系的不均匀沉积效应在反复的充放电过程中被不断放大,从而产生枝晶[8]. 同时,锂金属本身较低的库仑效应与逐渐增长的负极过电势也会导致整个体系容量降低[9].
固态电解质和锂金属负极组合既保障了体系的安全性,固态电解质相对优秀的机械性能与稳定的物化性质也使得其可有效抑制锂枝晶的生长,并提供了理想的循环性能与理论比容量,是下一代储能体系的理想之选. 但是,目前已报道的固体电解质中,依然没有一种单一的材料既能够提供足够宽的氧化还原窗口,使得其能够在锂负极与商用正极间的氧化还原电位间都保持稳定,又具有足够强的机械性能以阻挡锂枝晶的生长[10]. 目前,多数研究对于一种材料在电化学体系内的表现只关注单一性能的表征,较少将各种物理化学参数整合成一个可以量化的系统参与评估. 对进一步寻找合格的固态电解质材料造成了困难.
有限元仿真分析通过微分方程的数学近似,对真实的物理系统进行模拟,利用有限数量的组元对未知系统进行预测,在材料加工、机械制造、能源化工以及科学研究等领域均已广泛应用. COMSOL Multiphysics(简称COMSOL)作为一个成熟的跨平台有限元分析、求解以及多物理场仿真的软件. 其允许使用传统的基于物理学理论的用户界面以及偏微分方程,将体系内存在的多物理场进行耦合. 同时COMSOL也可对电池热管理进行评估,利用对于化学反应热的计算评估电池内部的冷却需求,避免在实际使用中过高的温度所带来的固体电解质界面膜(SEI)加快分解与气体析出. 同时由于电池内部工作时属于瞬态过程,故也可评估电池体系的热点和热梯度,以了解非均匀的温度可能导致的非均匀老化. 对于热启动管理的应用,可以进一步拓展到动力电池的热管理分析,研究汽车在冷启动时石墨上锂的析出和老化,以及低温对电解液传导的影响,评估锂电池包内部的温度分布. 因此仿真模拟对分析和预测锂电池的电化性能和安全管理具有重要的意义.
本文通过已经成熟的多物理场仿真软件,建立起电池体系内部传质过程、化学反应、电池内部极化、热效应与结构力学分布. 利用多物理场耦合,将这些同时发生的物理化学变化在一个模型内部同时考虑,通过稀物质传递模块改变化学反应的浓度梯度,将反应电流密度与实际反应的沉积量相耦合,从而了解在多物理场作用下电池体系内部的物理场与物质分布. 利用物理化学参数研究了一种较为成熟的LiPON型薄膜固态电解质相关的锂金属固态电池,所提出的建模思路可以为多物理场耦合的固态电池模型提供工作流程的指导.
1 仿真方法
模型的建立主要分为全固态锂电的材料和电化学过程的仿真及参与分析的热力学和力学场的仿真. 传统的锂离子电池在进行体系仿真时多采用均相Newman 多孔电极模型进行研究[11,12]. 因为传统锂离子电池的电解质采用液态电解质,表明其相关的化学反应以及传质过程均会在液态体系内进行,所以均质化多孔电极就很好地解决了固-液界面接触以及结构和颗粒的几何形状对体系性能的显著影响[13]. 但对于全固态锂电池,所有的界面都属于固-固界面,表明现有的多孔电极模型考虑固液相的有效接触并不能很好地解释固-固接触存在的问题. 所以,首先要给出全固态电池的理论模型.
1.1 全固态锂金属电池的材料仿真
模型讨论以钴酸锂(LCO)作为正极,以锂金属作为负极,以LiPON[LixPOyNz(x=2y+3z-5),本文采用Li3PO4]作为固体电解质的电化学体系的建模与相关参数. 分别讨论了在薄正极(面容量为210 mA·h·m-2,厚度为0.32 μm)与商用厚正极(面容量为30000 mA·h·m-2,厚度为45.77 μm)在不同条件下电池的电化学性能. 电池几何分别如图1所示. 其中,商用负极考虑大规模生产,与正极采用以1∶1的容量相比. 由于实验对于不同厚度的正极通常采用过量负极以保持一致性,本文采用与正极相同厚度的负极,方便模型建立.
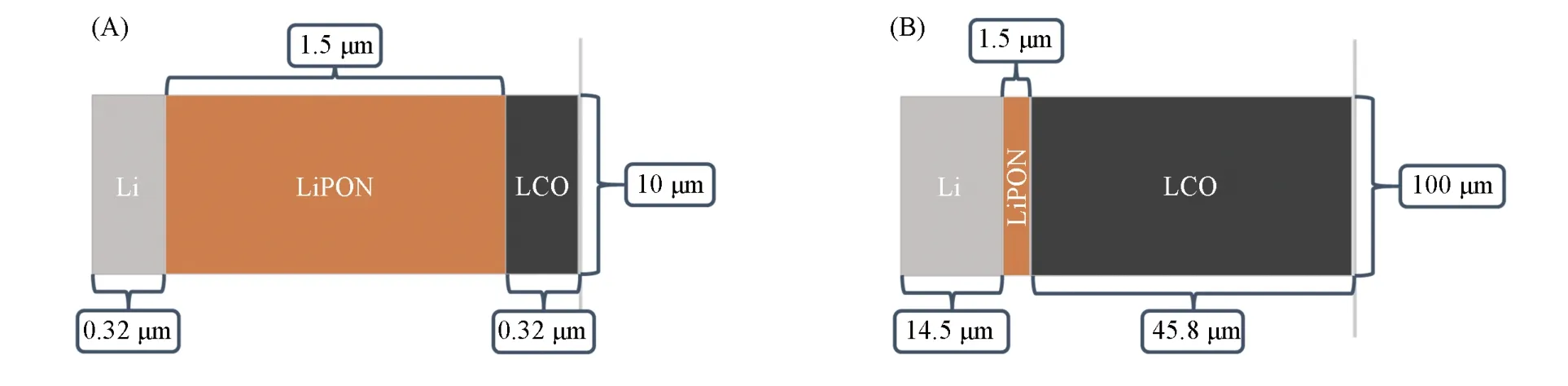
Fig.1 Geometry of thin cathode battery(A)and thick cathode battery(B)
1.2 钴酸锂正极的材料及电化学过程仿真
钴酸锂作为常见的商业正极材料,具有较高的氧化还原电位(工作电压达到4.2~4.5 V)与较大的实际比容量(约140 mA·h·g-1). 其相关的物理化学参数[14]列于表1.
钴酸锂对锂负极的平衡电位与平衡电位温度导数分别见图S1(A)和(B)(见本文支持信息)以插值函数的形式给出.

Table 1 Physical parameters of lithium cobalt cathode
由于钴酸锂正极与LiPON 薄膜以固-固接触的形式在固相界面发生反应,所以在实际模型中采用一个电极表面来仿真正极和薄膜固态电解质的界面,配合钴酸锂内部的稀物质传递,在瞬态模型中,不断将正极-电解质界面的锂离子/锂离子空位通过扩散传递入钴酸锂体相.
正极反应可用下列化学反应表示:
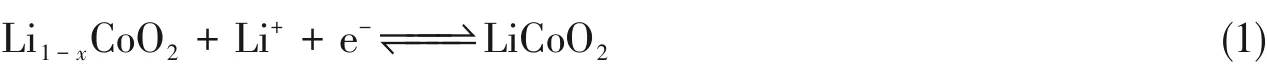
此反应可通过Butler-Volmer动力学方程描述[15]:
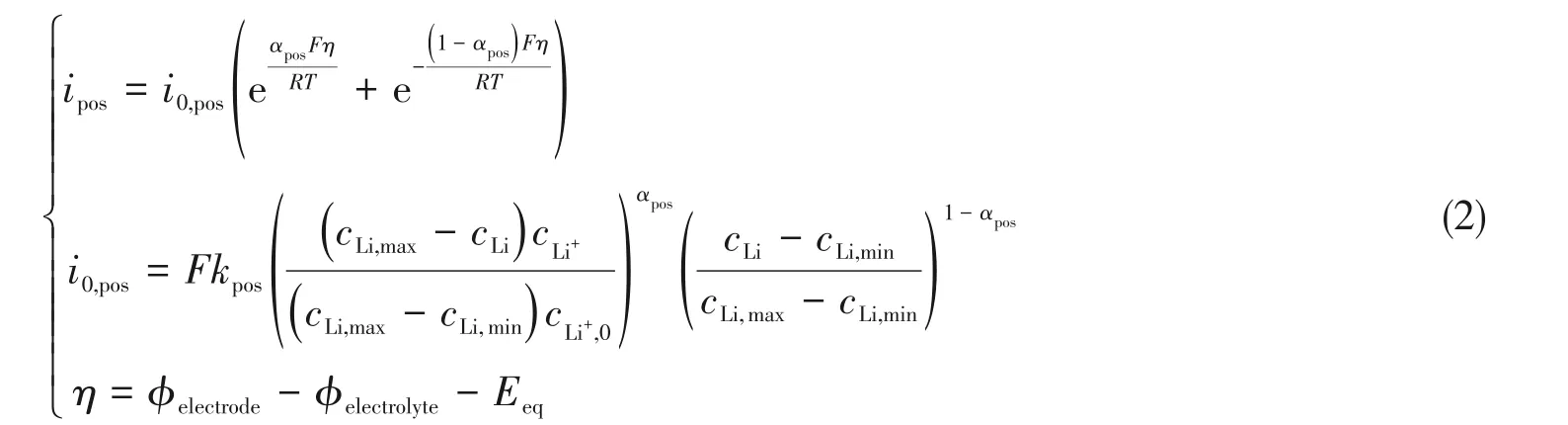
式中,ipos(A/cm2)为正极电流密度;i0,pos(A/cm2)为正极交换电流密度;αpos为正极反应的阳极传递系数;F(96485 C/mol)为法拉第常量;η(V)为过电势;R(8.314 J·mol-1·K-1)为气体常数;T(K)为温度;kpos(mol·m-2·s-1)为正极反应速率常数;cLi,cLi,max和cLi,min(mol/m3)分别为固体电极中锂的浓度、最大浓度与最小浓度;cLi+和cLi+,0(mol/m3)分别为正极与电解质锂离子的浓度;φelectrode和φelectrolyte(V)分别为电极与电解质的电化学势;Eeq(V)为平衡电位.

电极表面的反应符合下式:式中,ielectrolyte(A/cm2)为电解质内部电流密度;n为载流子荷电数,在锂电池内部为1;ielectrode(A/cm2)为电极内部电流密度;iloc,m(A/cm2)为电极反应内部电流.
在正极内部的稀物质传递使用菲克定律解决. 传质机理如下式:

式中,ci(mol/m3)为组分浓度;t(s)为时间;Ji(mol·s-1·m-2)为组分面通量;Ri(mol·s-1·m-2)为组分参与反应的速率;Di(m2/s)为组分的扩散系数. 同时,整个正极应该将除电极-电解质界面外的部分都设置为无通量的边界条件.
最后,将这个稀物质传递与电极表面进行电化学与电场的多物理场耦合,电极表面耦合的函数应该将化学反应的产物与电极的物质浓度相耦合. 耦合函数如下式:

式中,νi为化学反应当量系数.
1.3 锂金属负极材料及电化学过程仿真
锂金属负极作为金属电极,与石墨负极具有完全不同的反应机理. 石墨负极在充放电过程中锂离子不断在石墨结构内嵌入和脱出,锂离子不断通过扩散进入石墨体相内. 而锂金属负极在充放电沉积与脱嵌过程中完全可以看作锂离子在金属锂表面的反应,锂离子通过电解质在负极-电解质表面还原,并保持在负极-电解质表面上. 由于锂金属体相内部电荷守恒,锂离子不参与传质,且由锂单质构成,不存在其它物质参与物质传递,故锂金属负极体相内主要为金属内部空位扩散机理. 机理的不同简化了建模难度. 锂负极的物理性能参数[14]列于表2.

Table 2 Physical parameters of lithium anode
杨氏模量、泊松比、导热系数和等压热容在多物理场仿真时需要不同温度的精确值,故以插值函数的方式给出,见图S2(本文支持信息).
由于锂负极与LiPON 薄膜以固-固接触的形式在固相界面发生反应,所以在实际模型中采用一个电极表面来仿真负极和薄膜固态电解质的界面就可完成界面反应的仿真. 因为锂负极内部没有锂离子的扩散,所以并不需再像正极反应一样引入稀物质传递模块以讨论物质在电极内部的传递.
负极反应可用下列化学反应表示:

此反应可通过Butler-Volmer动力学方程描述:
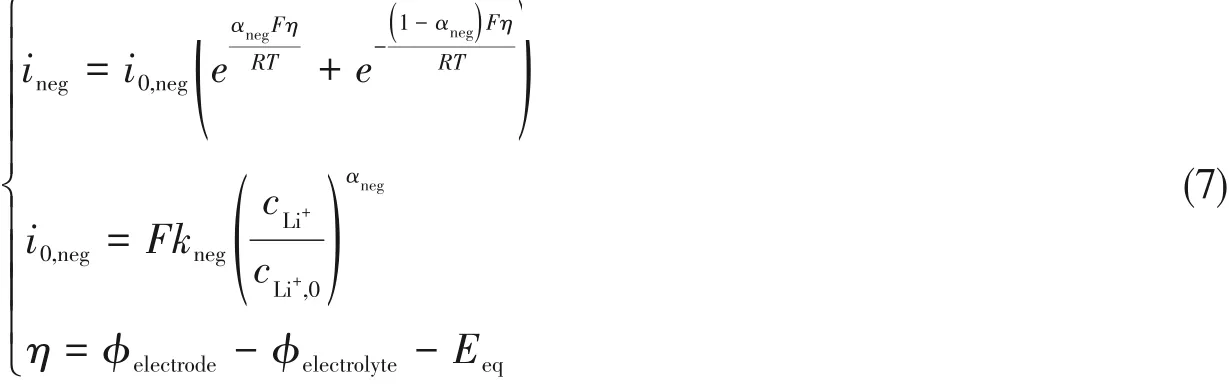
式中,ineg(A/cm2)为负极电流密度;i0,neg(A/cm2)为负极交换电流密;αneg为反应(6)的阳极传递系数;kneg(mol·m-2·s-1)为负极反应速率常数.
1.4 Li3PO4固态电解质的材料及电化学过程仿真
Li3PO4是一种典型的LiPON型固态电解质,Bates等[16,17]将其作为靶材,在N2气气氛中,采用射频磁控溅射的方法使其生长. LiPON具有可应用的离子电导率(约10-6S/cm)和极低的电子电导率(约10-12~10-14S/cm),对金属锂稳定,电化学窗口为5.5 V,是具有应用前景的薄膜固态电解质材料. LiPON是目前广泛用于微电池的固态电解质,是建模的理想选择. 表3列出了Li3PO4的物理化学参数[14].
Li3PO4所属的氧化锂-五氧化二磷(Li2O-P2O5)是一种典型的玻璃化系统. 通常,P2O5的准二维聚合物网络会在改性剂,如Li2O的作用下解聚[18]. 氧化锂导致五氧化二磷网络中的桥连氧原子转变为非桥连氧原子. 此“弱电解质”模型在玻璃化网络中会以两种形式存在,且LiPON特有的高离子电导是由内部的热力学上能量更高,更易发生迁移的非键合锂贡献的[19]. LiPON体系内氧键合锂(Li0)与自由移动的锂离子与之空位(Li+和n-)的相互转换过程如下:


Table 3 Physical parameters of LiPON[14]
式中,kd(s-1)为反应的分解速率常数;kr/(m3·s-1·mol-1)为逆反应反应速率常数.
自由移动的锂离子初始浓度cLi+,init(mol/m3)、电解质内锂离子总浓度cLi+,0(mol/m3)和空位初始浓度cn-,init(mol/m3)的关系如下:

式中,δ为平衡状态下可自由移动锂离子的分数;cLi0(mol/m3)为平衡时氧键合锂的浓度.
反应动力学参数之间的转化关系[14]如下:

由于固态电解质无法像液态电解质一样形成巨大的浓度差. 故整个电池体系的浓差过电位便位于电极表面附近;锂离子浓度显著降低,接近极限电流密度而产生. 所以,在考虑电流密度时,便不能只考虑欧姆极化与电化学极化,而必须要考虑到浓度过电位的影响. 因此,单纯的电流密度公式便不再成立,而要使用Nernst-Planck方程来描述[20].

式中,Ji(mol·m-2·s-1)为传质面通量;i(A/cm2)为面电流密度;zi为组分电荷数;φelectrolyte(V)为电解质电化学势.
在解决了电解质模型的传质机理后,还需设置边界条件. 与电极相垂直的两个面不但无物质传入,也没有电场极化,且不与导体相接触(否则会导致系统短路),故应设为绝缘边. 最后,将电解质内部的3次电流分布与电极两端的反应相耦合.
至此,完成了全固态锂电池两个电极及电解质共3个部分的传质及材料学的多物理场仿真.
1.5 固体传热与固体力学仿真
从固相反应与电化学的角度解决了全固态锂金属电池的体系仿真,但在实际研究过程中并不能只讨论电化学过程,在全固态电池的实际使用中,机械性能与热管理是两个研究重点. 电池在枝晶生长区的应力分布将在很大程度上决定锂枝晶生长后是否有足够的驱动力刺穿整个固态电解质,且应力分布的不均匀对于固相反应的能垒与界面阻抗都有极大的影响. 而热管理作为电池温度函数的分布,既体现了电池的能量转化效率,也一定程度上可用于评估电池在长周期循环中的安全表现,是否会存在热失控与在冷启动时是否会由于电极结构塌陷而导致体系加速老化.
1.5.1 固体传热 首先,考虑到动力电池包内,对于圆柱状电芯通常采用卷置的方式,故整个电池包与本文绘制的二维模型不同,两侧是外界气氛. 多数情况下,对于叠片电池,电芯两侧依旧是电芯,所以在考虑整个体系的热通量时,可以让外界温度与电池表面保持一致,而不是恒定不变,模拟得到的结果更接近实际运行时电芯的热力学环境. 将传热分为固体内部传热、固体表面对环境的热辐射以及体系内外的热通量3个部分[21].
对于固体内部传热,热传导方程如下:

式中,dz(m)为材料厚度;ρ(kg/m3)为材料密度;Cp(J·mol-1·K-1)为材料的等压热容;u(m/s)为速度矢量;q(J·m-2·s-1)为传导的热通量;Q(W/m3)为热源体积功率(如反应热、欧姆热);q0(J·m-2·s-1)为热通量;k(W/m)为导热系数;h(W·m-2·K-1)为传热系数;Text为外界温度.
同时,正负极与外界接触的表面还对外界有热辐射,固体表面热辐射的方程如下:

式中,ε为热辐射率;σ(kg/m2)为材料面密度.
在给定了传热与热辐射方程后,还需对整个模型做边界条件的限制,在实际电池中,电池的正负极极片面积远大于电池及电解质厚度,垂直于界面的上下边界(蓝色)比表面积较低,故电池纵向的热传递可忽略不计,可看作与外界热绝缘. 如图2所示. 同时将整个系统的初始值设为293.15 K,即电池运作的初始温度. 如此,建立了全固态电池内部的固体传热模型.
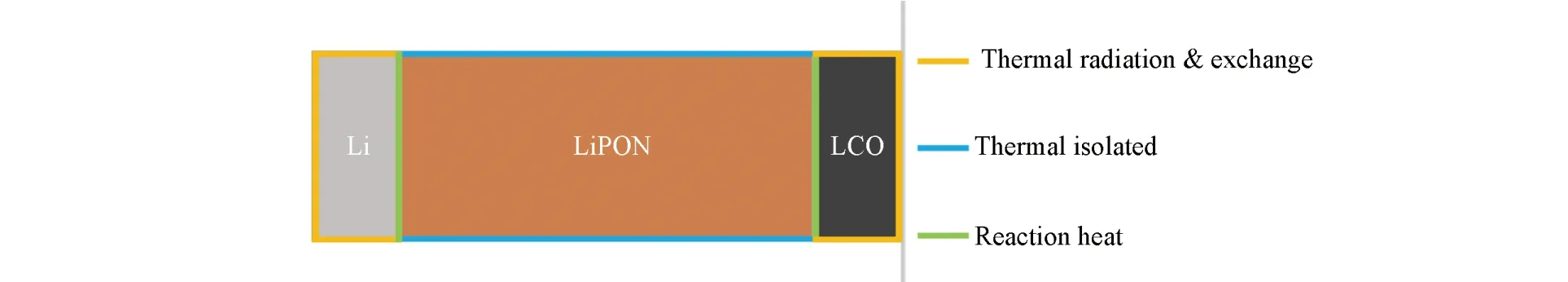
Fig.2 Boundary conditions of solid heat transfer
1.5.2 固体力学 采用已知数据研究锂金属内部的应力分布. 首先,将锂金属作为一种线弹性材料[22]. 对于固体力学内部线弹性材料的力学关系如下:

式中,F为形变梯度;S为形变率;I为等同张量;u(m/s)为位移场.
对于式(14)中的S,可以用下式计算:
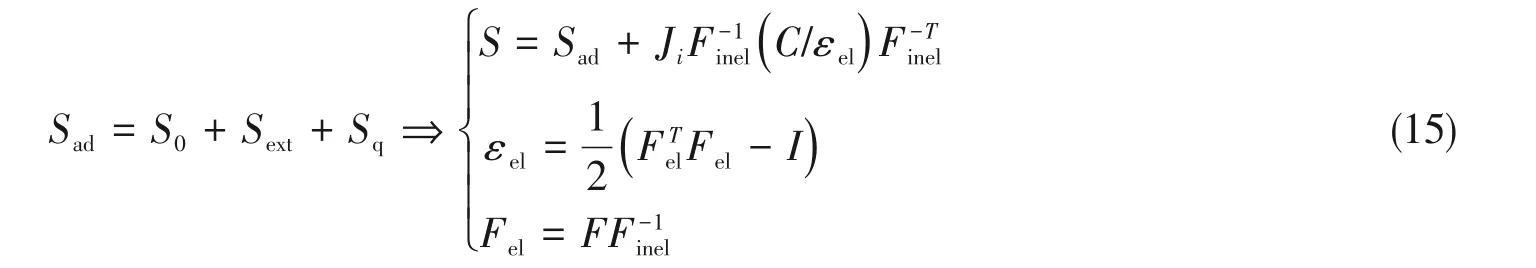
式中,Sad为总应变;S0为初始应变;Sext为外力应变;Sq为热应变;J为形变梯度的行列式;Finel和Fel分别为内弹性形变梯度和弹性形变梯度;C为右柯西-格林形变张量;εel为弹性工程应变张量.
工程应变张量ε和右柯西-格林形变张量可以用下面公式分别求出:
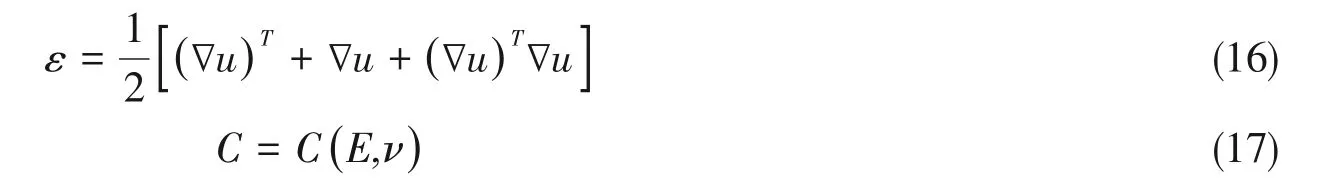
式中,E(Pa)和v分别是材料的杨氏模量和泊松比.
在给定了相关的物理化学方程后,需对模型进行边界条件的限制. 将锂负极作为研究主体,将锂负极与外界接触的面(蓝色)设定为自由面,将负极-电解质界面设定为固定约束. 指定锂负极的上下边为指定位移边,允许应力对其造成形变,如图3所示.

Fig.3 Boundary conditions of solid mechanical
1.6 求解思路
通过参数化扫描的功能对薄正极体系关于1.6C,3.2C,6.4C,12.8C,25.6C 和51.2C 等6 个倍率,对商业厚正极体系关于0.01C,0.02C,0.05C和0.1C等4个倍率分别求解,以得到不同放电倍率对不同正极所配合的固态锂电池负极金属锂生长的影响.
首先使用瞬态求解器求得体系的温度与电化学物质传递. 在放电结束后,得到各个时间的温度与之对应的沉积量和化学势等热力学信息. 通过体系的温度与最终沉积量,使用稳态求解器以得到电极的体积变化以及电极膨胀的比例,反推得到电极的应力分布.
2 结果与讨论
2.1 电化学性能
2.1.1 薄电极的仿真模拟 从薄电极首次放电曲线[图4(A)],可见,不同倍率下,随着放电电流强度增加,浓度过电势对过电势的贡献逐渐显著. 使得电池的电位提前到达放电终止电位,从而导致电池容量下降. 图4(B)给出了电解质内部锂离子浓度的分布也证明了此推论. 可见,不同倍率下放电结束时锂离子分布随着电流强度逐渐增大,大电流产生的浓度过电势导致了更加陡峭的物质分布.
在1.6C倍率下,随着放电的不断进行,电解质电势在放电初始态仅有电解质本身的欧姆极化做贡献,故呈斜率较小的线性关系[图4(C)]. 随着反应的不断进行,正极-电解质界面周围在电解质内部的锂离子被不断消耗. 正极内部的锂离子浓度却因为反应的进行不断增加,且锂离子在正极内部的扩散过程无法第一时间将锂离子从界面传导入整个正极. 从而在该界面附近造成了更大的浓度过电势,所以,在放电中程,电解质电势在靠近正极的部分降低更快. 而随着反应的进一步进行,整个电解质内部的浓度梯度又回归到稳态传质的线性分布,电解质的电位降就又呈现为线性降低,但是由于浓度过电势的客观存在,故斜率比起反应开始时大很多. 由图4(D)可见,在放电终止时,6.4C以下的倍率,电解质内部的电位降都较好地呈现线性分布,但是随着倍率的不断提高,反应时间缩短,在高倍率(特别是25.6C和51.2C)下,在放电结束时,整个电池的电解质内部无法达到稳态. 使得靠近正极-电解质界面的锂离子浓度降比电解质中其它位置更严重,也就导致在界面附近更大的电位降. 通过电位降可很好地了解浓度过电势对于电池容量及性能造成的影响.
从电解质电位降的角度解释了固态锂电内部的极化后,再从正极锂离子浓度的角度了解电池内部的物质分布. 从图4(E)可以看出,随着倍率的不断提高,放电时间缩短,正极锂插层反应速度加快,正极内部的稀物质传递速率逐渐无法在放电结束时维持稳态. 使得锂离子在接近正极-电解质界面处堆积. 物质分布不均,也在一定程度上增大了正极表面与电解质之间的浓度差. 由图4(F)可见,随着时间的推进,正极内部不只总体浓度增加,正极-电解质界面附近的浓度也比靠近集流体一侧增长速度更快. 表明电极内部的扩散传质无法满足大电流的锂离子嵌入.
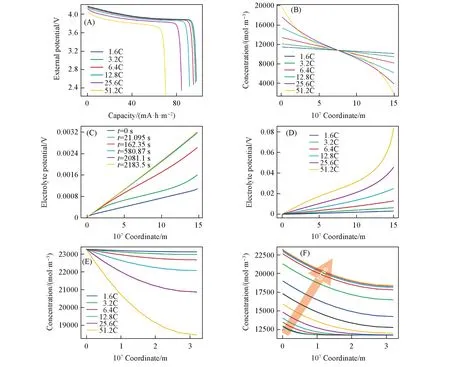
Fig.4 Discharge curves for thin cathode(A), concentration of Li ions in electrolyte at the end of discharge(B),potential of different sites in electrolyte 1.6C(C), the end of discharge for thin cathode(0 sets as the boundary between Li anode and LiPON)(D), Li ion concentration in thin cathode at the end of discharge(E)and Li ion concentration at different time of 51.2C discharge rate in thin cathode(F)
在分析了正极与电解质的锂离子分布后,将不同倍率下放电结束时在正极与电解质内部的锂离子分布同时观察(图5). 可见在正极-电解质表面存在一个非常大的浓度阶跃,且在大电流密度下更加明显. 同时,可见较大的浓度梯度不仅在正极-电解质界面能够观察到,在负极-电解质界面,大电流下同样存在显著的浓度梯度. 这是由负极锂与电解液中锂离子的快速传质与交换所致.
虽然在大电流分布下,浓度过电势对体系总过电势的影响起了绝对主导,但还需比较电池内部欧姆极化、电化学极化和浓度过电势对电池内部极化的贡献率. 从图S3(见本文支持信息)可见,随着正极表面锂离子由于大电流而堆积,正极表面浓度过电势先由于电化学反应的过快发生,电化学极化增强,导致浓度过电势对于体系的总极化贡献减小而减小,后由于正极内部扩散限制,锂离子难以插层堆积在正极-电解质表面导致浓度过电势迅速增加. 由于kneg=10-2mol·m-2·s-1,kpos=5.1×10-4mol·m-2·s-1≪kneg,故负极的电化学极化对过电势几乎无贡献,与文献结果相吻合[23,24].
2.1.2 商用正极的仿真模拟 相比薄电极,商业正极的负载更大. 在传统的液态电池中,液体电解质往往浸润入正极孔隙,参与正极内部传质. 液相的传质作用对于薄正极而言并不起决定性作用. 但是对于商业的厚正极,其厚度是本文仿真的薄正极的百余倍(45.8 μmvs. 0.32 μm). 可见,没有液体电解液的参与,正极内部扩散传质极慢,导致了固态锂电池厚电极电化学性能的降低. 从图6可见,在极低的放电倍率(0.01C)下,直接采用商用正极也只释放约5%的理论总容量. 可以推测,在无液态电解质存在下,正极内部固相传质成为影响整个体系运行的一大困境. 然而,对于目前多数的固态电解质体系,很难应用液态电解液采用的直接浸润入正极内部的传质策略. 从容量的释放比例来看(倍率与容量释放比例基本成反比),由于固相内部的传质速率远低于电化学反应速率,成为唯一的控速步骤.
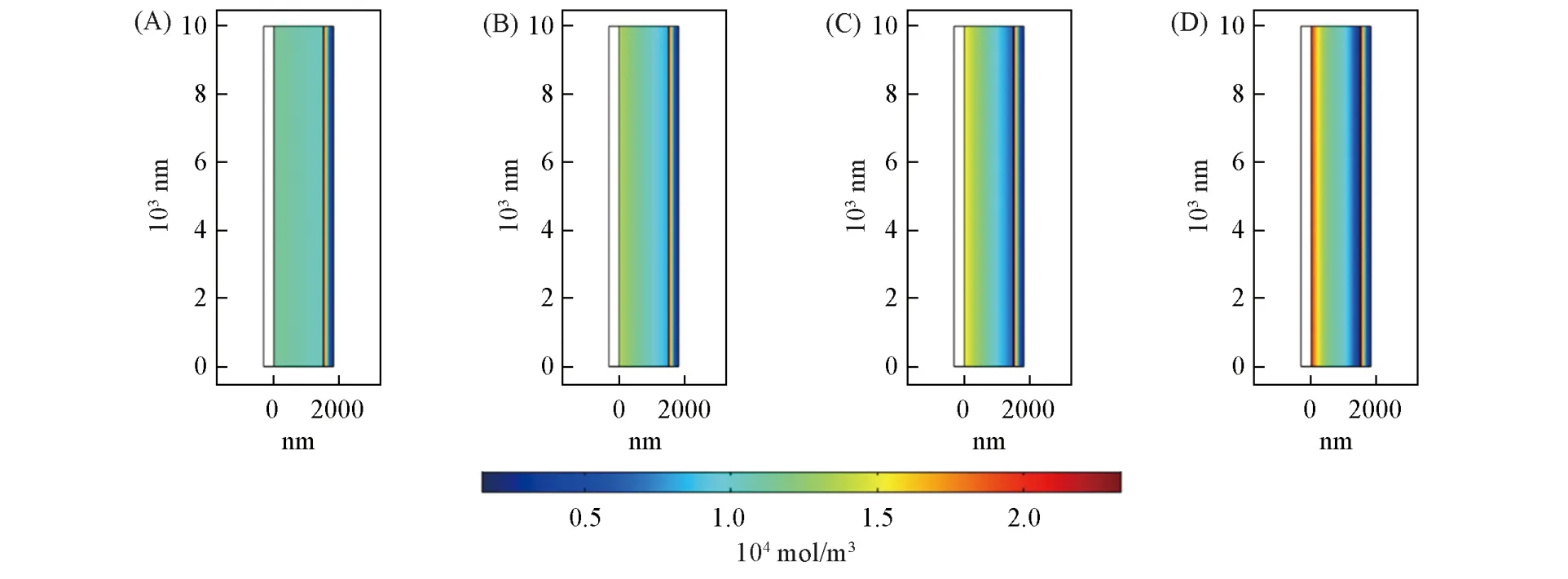
Fig.5 Li ion concentration of electrolyte and thin cathode at the end of 1.6C(A), 6.4C(B), 12.8C(C)and 51.2C(D)discharge
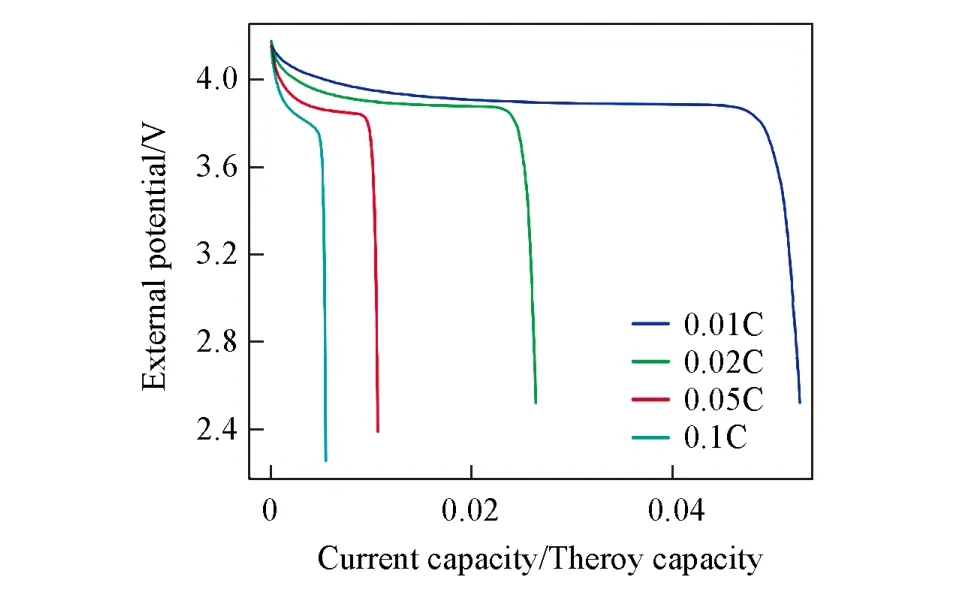
Fig.6 Discharge curves of commercial cathode
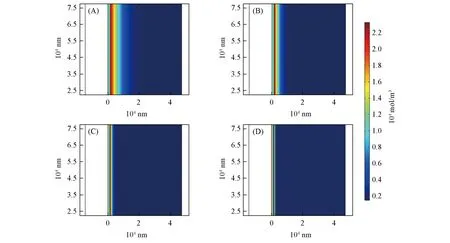
Fig.7 Li ion concentration of electrolyte and commercial cathode at the end of 0.01C(A), 0.02C(B),0.05C(C)and 0.1C(D)discharge
将不同倍率下放电结束时在商用正极与电解质内部的锂离子分布结合(图7). 可见,随倍率提高材料内部多数锂空位由于扩散步骤的限制,无法为参与电化学过程的锂离子提供反应位点. 在较低的倍率(0.01C)下,也只有表面6~7 μm的材料实际参与到了电化学反应中. 对厚度达到45 μm的正极远远不够. 此现象在大电流下更加明显. 与薄电极不同的是,较大的浓度梯度并不存在于负极-电解质界面处,电解质内部锂离子浓度分布基本趋于一致.
由图S4(见本文支持信息)可见,不论是在0.01C倍率还是0.1C倍率下,正极内部浓度过电势对电池内部极化贡献均最大. 电流大小对贡献占比基本没影响,正极内部的传质已经成为控速步骤. 目前商用正极无法与固态电解质直接组合,以提供能够满足实际使用倍率的充放电性能与传质速率.
2.2 温度变化和应力分布
2.2.1 薄正极的仿真模拟 在关于温度的研究中,主要关注电池的放电截止温度与整个放电过程的温度变化,从表4和图S5(见本文支持信息)可见,在倍率<12.8C时,由于体系倍率的提高,内部极化增加,同时放电时间缩短,导致在终止放电时体系温度逐渐升高. 而倍率>12.8C时,由于体系极化过于严重,导致放电过程提前截止,有大量的化学热无法释放,导致体系截止温度又转而下降. 而从图S6(见本文支持信息)体现的温度变化曲线也可见,整个体系的温度变化速率与体系的极化程度存在一定关系,这也是在51.2C倍率下,放电过程中电池体系温度先缓慢增加而后急剧上升的原因.

Table 4 Final temperatures for different C-rate on average
在关于应力场的研究中,同时考虑了温度不同导致的热膨胀、体积变化以及非均匀沉积对于整个体系应力分布的影响. 从图8可见,随着充电过程的倍率升高,负极表面沉积受动力学控制而变得不均匀,同时温度逐渐增加. 在12.8C~25.6C 倍率之间,充放电体积变化后无法进行有效的应力释放.同时,由于沉积的不均匀,大电流作用后,体系由于动力学因素占主导,从而有更强的应力紧张,从而导致更强的压力分布,体系的内部应力到达顶峰. 而后由于电池内部极化变大,充放电过程均提前终止,使得电池在沉积过程中的体积变化不显著,而导致电池体系应力有所降低. 这也可见,锂金属电池枝晶在大电流下易刺穿体系,不仅是锂的无序化沉积本身造成的,电池本身有限的空间结构也有一定贡献,但是这一空间限制很可能由于大电流导致充电容量无法完全发挥,从而变得不显著. 由图8还可见,边界的应力分布由于存在移动边,存在释放应力的可能,从而相比锂金属负极的中心更小.可以考虑应用一定的电池结构设计,将更多的应力提前分布在边界,从而降低整体的紧张. 在本文的模型中,将锂负极-电解质界面设为固定边,在实际体系中,锂金属负极的沉积/剥离也会导致固体电解质内部的应力聚集,会引发固体电解质,尤其是无机固体电解质产生裂纹,从而引发自发恶化,导致固体电解质的失效.
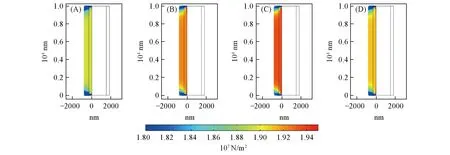
Fig.8 Distribution of stress in the end of deposition for 1.6C(A), 6.4C(B), 12.8C(C) and 51.2C(D)in thin cathode system
2.2.2 商业正极的仿真模拟 对于商用正极温度的研究,由于整个体系的反应热基本仅由体系容量决定. 从表5和图S7(见本文支持信息)可见,随着放电容量的骤减,整体的温度变化也随之下降. 由于商用正极与LiPON配合后总体容量释放都比较低,故总体温度变化比薄电极小很多.

Table 5 Final temperatures for different C-rate on average in commercial cathode
在应力场的研究中,商业正极也由于其较低的容量释放,与薄正极之间产生了完全不同的结果.从图9可见,由于电极的有效容量随着倍率变大而骤减,带来的体积变化基本成为影响电极应力分布的唯一要素. 总体应力由于容量减小而带来的体积变化骤降. 可见,在0.01C倍率下,负极充电结束后整体控制在5 MPa左右;而当倍率>0.1C时,负极的应力由于体积变化减少而骤减至0.7 MPa左右.与商用正极的结果一致的是,边缘由于其具有更大的自由度从而相对具有较小的应力分布. 说明对于锂电池的负极体系设计可以采用类似的策略.
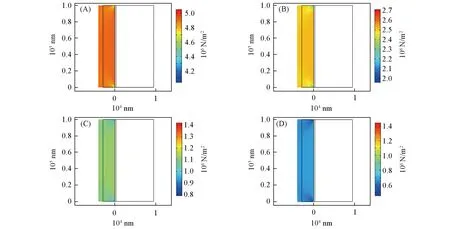
Fig.9 Distribution of stress in the end of deposition for 0.01C(A), 0.02C(B), 0.05C(C) and 0.1C(D)in commercial cathode system
3 结 论
基于有限元多物理场仿真软件,对于LiPON基全固态锂金属电池体系进行了仿真. 利用3次电流分布完成了固体电解质传质模型的建立;利用电极表面仿真锂负极的反应表面,再利用稀物质传递加上电极表面模块的耦合完成了整个电池电化学体系的构建. 结合固体力学与固体传热进行多物理场耦合,完成了对于体系的应力以及温度的仿真与监控.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200451.
猜你喜欢
起重运输机械(2023年22期)2023-12-12 09:55:52
小读者·爱读写(2023年9期)2023-10-02 03:46:59
小读者(2023年18期)2023-09-27 04:38:38
陶瓷学报(2021年1期)2021-04-13 01:33:40
安徽电子信息职业技术学院学报(2019年2期)2019-04-26 06:38:28
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中学生数理化(高中版.高二数学)(2017年1期)2017-04-16 05:33:49
制造业自动化(2017年2期)2017-03-20 14:26:08
中国塑料(2015年6期)2015-11-13 03:03:05
电源技术(2015年9期)2015-06-05 09:36:06
