
基于刚毛柽柳转录组测序的EST-SSR标记识别与开发
2021-05-12 14:09:02李佳彬张雅楠贾媛媛田芸芸党振华
草业科学 2021年4期
李佳彬,黄 蕾,张雅楠,贾媛媛,田芸芸,张 雷,党振华
(1. 内蒙古大学生态与环境学院 / 蒙古高原生态与资源利用教育部重点实验室 / 内蒙古草地生态学重点实验室,内蒙古 呼和浩特010021;2. 内蒙古大学生命科学学院 / 牧草与特色作物生物技术教育部重点实验室,内蒙古 呼和浩特 010021;3. 内蒙古自治区林业科学研究院,内蒙古 呼和浩特 010010;4. 内蒙古大青山森林生态系统定位观测研究站,内蒙古 呼和浩特 010010)
刚毛柽柳(Tamarix hispida)是中亚的广布木质盐生植物,是柽柳科(Tamaricaceae)柽柳属(Tamarix)最耐盐碱的种类之一,常分布于我国荒漠或半荒漠地带的盐土或低湿沙区[1]。它不仅在水土保持、防风固沙和绿化造林方面具有重要的生态价值,丰富的营养与化学成分还赋予其很高的药用和经济价值[2-3]。目前对刚毛柽柳的研究主要集中在形态解剖[4]、群落生态[5]、生理生态[6-7]、植物化学[8]、分子生态[9-10]等方面。然而,作为荒漠地区的主要资源树种,对其适应生境的内在分子基础还知之甚少,亟需深入了解和研究,以进一步挖掘蕴藏在该物种中的特有遗传资源。
SSR (simple sequence repeat),即简单序列重复,指重复基序为1~6 个核苷酸的串联重复序列,广泛分布于真核生物基因组中,每隔10~15 kb 就存在1 个SSR 位点[11]。目前,SSR 分子标记已被用于遗传图谱的构建、遗传多样性分析、品种鉴定及分子育种等研究领域[12-14]。依据SSR 在基因组中的分布,可将其分为基因组SSR 和表达序列标签SSR(EST-SSR)[15]。基因组SSR 位于基因组非编码区,通常情况下,等位基因数目及多态性均相对较高,而EST-SSR 由于位于基因编码序列内部,具有相对保守的特点。大量研究表明,EST-SSR 的变异能改变基因的活性或调节基因表达,故常与基因的功能密切相关,可影响相关的生物和细胞过程,如蛋白质结构、传感和信号传递及基因转录等[16-17]。例如,编码区(CAG/CTG)n 重复的扩增或减缩可导致蛋白质错误折叠或基因表达异常[18];(CCTG/CAGG)n 重复与复制起点的距离可能在决定这些重复序列的遗传不稳定性方面发挥重要作用[19];(GAA/TTC)n 重复可形成三聚体,相关结构被称为“粘性DNA”,在模板上捕获RNA 聚合酶,从而阻断转录延伸[20-21]。因此,EST-SSR 可能是生物体适应环境变化的分子装置[22],鉴定和深入分析多态EST-SSRs 的功能可为探索植物适应性进化机制提供新见解[23-24]。
CandiSSR 是一款近年开发的多态性微卫星识别软件,可同时比较相同或近缘物种多个样本的基因组或转录组序列,进行大规模多态性SSR 的识别及分析[25]。本研究通过转录组测序构建了5 个不同样地刚毛柽柳的基因序列集,利用CandiSSR 软件系统识别了该物种多态EST-SSR 位点;随后对所识别多态SSR 的数目、类型、出现频率、基因中的位置及关联基因的功能进行了全面分析;经验证,从15 个随机挑选的SSR 中共开发出13 个多态性SSR 标记。本研究可为进一步鉴定由EST-SSRs 变异驱动的该物种生境适应机制提供素材,为开展其遗传多样性评价和种植资源开发奠定基础。
1 材料及方法
1.1 植物材料
2019 年9 月,在内蒙古阿拉善盟额济纳旗采集了5 个样地刚毛柽柳(Tamarix hispida)的幼嫩叶片,详细采样地点的地理信息如表1 所列。每个样地中每隔5~10 m 采集20~24 个个体。样本采集后装入冻存管,并迅速置于液氮速冻,带回实验室备用。
1.2 试验方法
1.2.1 总RNA 提取及质量检测
每个样地选择1 株植物用于总RNA 的提取。使用RNA plant plus 植物总RNA 提取试剂(DP437,天根生化科技有限公司,北京)进行。利用1%琼脂糖电泳和超微量紫外分光光度计(NanoDrop 2000c,赛默飞世尔,美国)检测总RNA 的质量。检测合格后的RNA 样本置于干冰中寄送至安诺基因(安诺优达基因科技有限公司,北京)。

表 1 5 个刚毛柽柳种群的地理位置信息Table 1 Location information for five Tamarix hispida populations
1.2.2 cDNA 文库构建及转录组测序
利用安捷伦2100 RNA Nano 6000 Assay Kit (Agilent Technologies, 美国) 对RNA 样本进行完整性检测,RNA 完整值(RNA integrity number,RIN)达到7.0 以上。然后由测序公司进行测序文库构建,简要流程为:用带有Oligo(dT) 的磁珠富集mRNA,向得到的mRNA 中加入片段缓冲液使其成为短片段,再以片断后的mRNA 为模板,用六碱基随机引物合成cDNA第一链。构建好的文库利用Illumina HiSeqTM4000测序平台进行测序。
1.2.3 转录组de novo 组装及注释
对原始序列进行过滤得到质量较高的Clean Reads,随后采用Trinity (v2.4.0)分别对5 个样本的Clean Reads 进行de novo 组装,得到每个样本转录组的Unigenes,然后进一步将各转录组Unigenes 进行拼接和去冗余,得到非冗余All-Unigenes。通过Blast、HmmScan、SignalP、TmHMMP 等 工 具 对All-Unigenes 进行功能注释,采用Trinotate (v3.0.2)整合功能注释信息。利用TransDecoder (v3.0.1)对unigene的编码区进行鉴定,确定蛋白质编码序列(CDS)。
1.2.4 多态SSR 位点识别与定位
利 用 CandiSSR (https://github.com/xiaenhua/CandiSSR)识别5 个刚毛柽柳转录组中的多态性SSRs[25]。筛选标准:包含2、3、4、5、6 重复单元的SSR 至少出现次数分别是6、5、5、4 和4 次。对于具有全长CDSs 的Unigenes,可根据SSRs 与相应基因起始(ATG) 和终止(TAA、TAG、TGA) 密码子的相对位置来分析SSRs 的位置。对于组装为非全长CDSs 的Unigenes,通 过BLAST(http://blast.ncbi.nlm.nih.gov/Blast.cgi)识别Genbank 中的全长同源基因,然后根据其在查询序列中的位置预测SSRs 的位置。对于识别出的SSR,采用Primer 3.0 进行引物批量设计,设计原则:①引物长度18~24 bp;②退火温度在53~62 ℃;③PCR 产物长度100~200 bp;④GC含量在40%~60%。
1.2.5 DNA 提取及多态SSR 标记开发
利用植物基因组DNA 试剂盒(DP305,天根生化科技有限公司,北京)从30 个刚毛柽柳样本中提取基因组DNA,并用1% 琼脂糖凝胶电泳和NanoDrop 2000c (赛默飞世尔,美国)进行DNA 质量及浓度的检测。随机挑选15 个多态SSR 位点进行验证,PCR 扩增引物由擎科新业生物技术有限公司(北京)合成。
使用ABI2720 PCR 仪进行PCR 扩增,选择25 μL反应体系:50 ng·μL-1DNA 模板1 μL,12.5 μL Premix Taq (宝生物工程有限公司,大连),上、下游引物各0.5 μL(10 μmol·L-1),10.5 μL 的ddH2O。PCR 扩增程序:94 ℃预变性5 min;94 ℃变性30 s,退火30 s,72 ℃延伸30 s,30 个循环;72 ℃ 10 min 终止反应。扩增产物用2% 琼脂糖检测。对于扩增成功的引物,在5′端加入3 种荧光染料(6-carboxy-fluorescine,hexachloro-6-carboxy-fluorescine,6-carboxy-X-rhodamine)中的一种进行标记,然后用相同的PCR 程序对所有检测个体再次扩增,结果在ABI 3 730 DNA 分析仪(Applied Biosystems,北京) 上分析,毛细管电泳的内标为GS500LIZ。从上述验证成功的引物中,选择5 对引物(TR25868、TR19384、TR18283、TR23634、TR23597)的PCR 产物,依次进行2% 琼脂糖凝胶电泳、凝胶回收及DNA 片段纯化(爱思进生物技术有限公司,杭州),将回收产物与pMD19-T 载体(宝生物工程有限公司,大连)连接并转化大肠杆菌(全式金生物技术有限公司,北京),经菌液PCR 鉴定后,选取阳性克隆送至擎科新业生物技术有限公司(北京)进行测序。
1.2.6 数据处理
采用GeneMarker (v2.6)对毛细管电泳峰图进行基因型判读及基因型统计,每个位点用GenAlEx(v6.5)对等位基因数(Na)、观测杂合度(Ho)和期望杂合度(He)进行计算,然后用PowerMarker (v3.0)测量多态性信息含量(PIC)。通过GenePop (v4.7)查看各位点是否偏离哈迪-温伯格平衡和连锁不平衡。
2 数据分析
2.1 测序和de novo 组装结果
经测序,5 个样本的Clean Reads 最小为44 180 102条,最大47 575 708 条,平均Q30为93.13%。对上述Clean Reads 进行组装后,每个个体分别获得超过31 000 条Unigenes,平均长度范围在1 043.24~1 139.67 bp,平均N50为1 913~1 970 bp (表2)。进一步组装和去冗余后,共得到72 661 条All-Unigenes,总长度为65 674 878 个核苷酸,平均长度为903.85 bp,平均GC 含量为40.62%,N50长度为1 578 bp。
2.2 多态SSRs 的识别
在5 个刚毛柽柳转录组中,共鉴定到1 187 个多态性SSRs 位于1 123 个基因序列中,共有154 个SSR 重复单元类型,二、三、四、五、六核苷酸重复出现频率有较大差异(图1)。最常见的SSRs 是三核苷酸重复类型,共646 个(54.42%),最丰富的重复序列类型有AGC/GCT(105,16.25%)、AAT/ATT(89,13.78%)、AAG/CTT (85,13.16%)等;其次是二核苷酸重复,有447 个(37.66%),主要基元以AG/CT (217,48.55%)和AT/AT(177,39.60%)为主;四、五、六核苷酸重复类型的数量较少,占总SSRs 的7.92%,3 种重复类型分别以ATAA/TTAT(8,22.86%)、AAATA/TATTT(8,33.33%)、CCCTCT/AGAGGG (5,14.29%)为主。
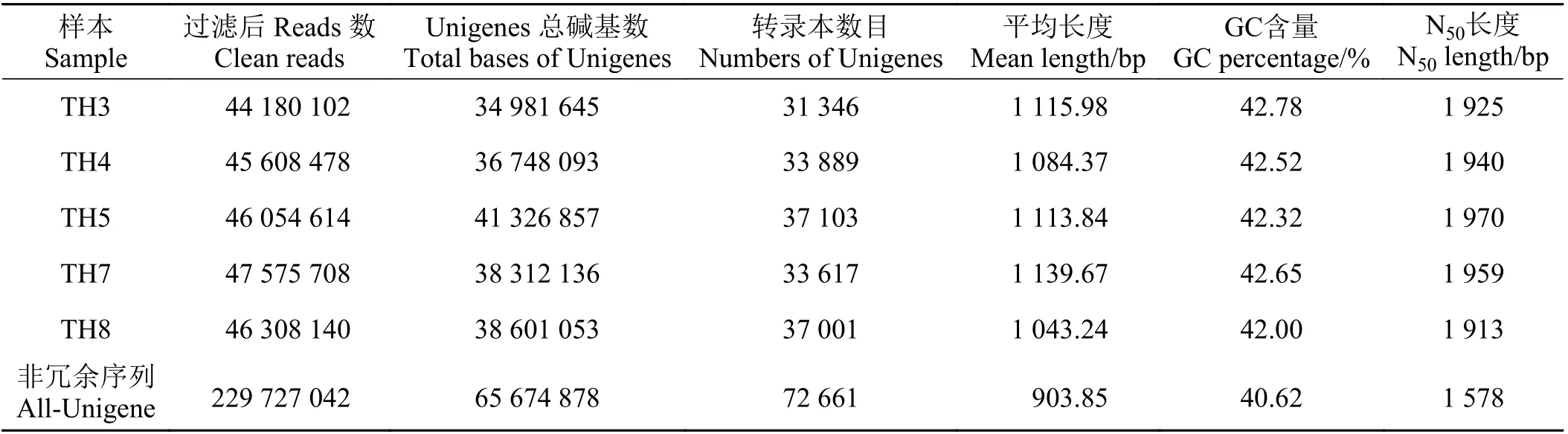
表 2 测序数据产出与组装结果Table 2 Sequencing outputs and assembly results

图 1 SSR 重复类型及频率示意图Figure 1 Diagram of SSRs' motif types and frequencies
2.3 SSR 位置预测及频率分析
在多态SSR 对应的Unigene 中,有856 个与公共数据库中的已知蛋白质有同源比对结果。887 个SSRs 分别位于829 个Unigenes 的编码区(CDSs)和非翻译区(UTRs) 内。其中500 个位于CDSs 中,176 个位于3′UTRs,211 个位于5′UTRs。三核苷酸重复序列(91.40%)多位于CDSs 区,AGC/GCT 相对丰富;二核苷酸多位于UTRs,在3′UTRs 中占61.93%,其中AT/AT 相对丰富;5′UTRs 中占62.56%,AG/CT相对丰富(图2)。

图 2 SSR 分布分析图Figure 2 Simple sequence repeat distribution analysis diagram
2.4 多态SSR 关联基因的GO 及KEGG 功能注释
GO 功 能 注 释 显 示,含685 个SSRs 的635 条Unigenes 与GO 数据库中的5 582 个功能基因有比对结果(图3)。它们被划分为3 大类,分别为2 428个生物过程(43.50%)、1 253 个细胞组分(22.44%)和1 901 个分子功能(34.06%)。进一步将三大功能细分为1 248 种亚类。其中,生物过程包括743 个功能亚类。“调节转录”(regulation of transcription)类别在3′UTRs 与5′UTRs 内含有SSR 的Unigene 中所占比例最大,分别占GO 注释总Unigene 的5.82%和8.27%;“转 录”(transcription) 类 别 在CDSs 含 有SSR 的Unigene 中最多(占10.17%);细胞组分包括161 个功能亚类,代表性功能为“细胞核”(nucleus),分别在3′UTRs、5′UTRs、CDSs 中 含 有SSR 的Unigene 中占20.59%、19.01%和29.77%;分子功能包括344 个功能亚类。“转录因子活性”(transcription factor activity)类别在3′UTRs (10.63%)及CDSs (13.99%)含有SSR的Unigene 中 最 多,“ATP 结 合”(ATP binding)在5′UTRs (8.80%)含有SSR 的Unigene 中最多。
KEGG 注释分析显示,120 条含有多态SSR 的Unigenes 参与到110 个通路中。对于包含多态SSR的5′UTRs、CDSs 和3′UTRs,参与最多的是“代谢途径”(metabolic pathways),分别在5′UTRs、CDSs 和3′UTRs 含有SSR 的Unigene 中占13.08%、12.60%和11.76%。其次是“植物激素信号转导”(plant hormone signal transduction),分别在5′UTRs、CDSs 和3′UTRs含有SSR 的Unigene 中占8.41%、11.02%和7.06%。
2.5 多态性SSR 引物筛选与标记开发
利用1 187 个多态SSRs 的侧翼序列,成功为1 182 个SSRs 设计出了PCR 扩增引物。在随机选取的15 个多态SSRs 引物中,13 个成功扩增出多态位点(表3)。为进一步验证多态SSR 的可信度,选择上述成功扩增的5 个位点,分别进行了SSR 片段的回收、克隆及测序。结果如图4 所示,测序结果(图4C)与基于高通量测序组装的基因序列(图4A)完全吻合,且利用CandiSSR 识别到的SSR 重复单元变异与毛细管电泳检测到的多态性一致(图4B)。同时发现,个别SSR重复内存在碱基替换事件,例如:TH7_DN13421、TR_25868_G7:A→C(图4A);TR_25868_G16:G→C(图4C)。

图 3 含有多态SSR 位点基因序列的GO 功能注释结果Figure 3 Results of the gene ontology function annotation of the polymorphic SSR-containing sequence
在这些SSR 中,共检测到73 个等位基因,每个位点得到3~8 个等位基因(平均5.615 个)。期望杂合度(He) 的范围为0.301~0.786,观测杂合度(Ho)的范围为0.133~0.800,均值分别为0.619 和0.476。多态性信息含量(PIC)值范围为0.283~0.744,平均值为0.565 (表3),所有位点均符合哈迪-温伯格平衡(P > 0.05),且未检测到连锁不平衡现象。
3 讨论
高通量测序技术极大推进了获得非模式物种基因序列的速度。利用这些数据,再结合一些生物信息学软件(MISA[26]和SSR Finder[27]等),研究人员可快速识别出相应物种数以千计的SSR 位点。无论开展种群遗传学研究还是对特定SSR 的功能进行分析,首先都要得到一组多态性的SSR 标记。然而,对基于高通量测序数据的海量SSR 鉴定多态性,仍然是该标记技术应用和研究的瓶颈[24]。本研究通过构建相同物种不同分布区个体的转录组数据集,利用CandiSSR 在5 个刚毛柽柳转录组中共识别出1 187个多态性SSRs,随机挑选的15 对引物中有13 个为多态性SSR 标记,成功率近87%,大大提高了该物种多态性SSR 标记开发的效率。遗传参数分析表明,这些SSR 的平均PIC 为0.565,平均He 为0.619,平均Ho 为0.476,呈现高度多态性,可用于该物种后续的种群遗传学和适应进化研究。同时,本研究所识别的SSR 数目高于用相同软件鉴定的南北两地区银缕梅(Parrotia subaequalis)的497 个SSRs[28]、四合木(Tetraena mongolica)的881 个SSRs[29],低于茶树(Camellia)鉴定的1 663 个SSRs[30],一定程度反映了该物种SSR 变异相对丰富及不同物种SSR 含量的差异。当然,这一差异也可能是因检测个体数、识别参数不同造成。在刚毛柽柳多态SSR 中,三核苷酸出现频率最高(46.93%),且91.40%的三核苷酸重复位于CDSs 中,这与荔枝(Litchi chinensis)和刺梨(Rosa roxburghii)的研究结果相符[31-32]。这可能是因为密码子由3 个核苷酸构成,若在编码区发生突变,将引起最轻的蛋白质序列突变[33]。在本研究中,CDSs 中的SSRs 表现出对三核苷酸的强烈偏倚性,最丰富的三核苷酸重复为AGC/GCT,其次是AGG/
CCT、ACC/GGT、ATC/GAT 和AAG/CTT。这与Sonah等[34]认为的AAG、AAC、ATC、AGC、AGG 和ACG重复是双子叶植物外显子中最常见SSR 类型的观点一致,一定程度反映了本研究SSR 频率分析的正确性。

表 3 开发多态EST-SSR 标记的信息Table 3 Detailed information of the developed polymorphic EST-SSR markers
刚毛柽柳生态幅较广,不同分布区的水分、温度、地形、土壤属性等均存在较大差异,不同生境下的种群很可能进化出不同的适应机制。张娟等[35]用随机扩增多态性DNA (random amplified polymorphic DNA, RAPD)方法描述了分布在新疆的9 个刚毛柽柳居群的遗传分化及遗传结构;张道远等[5]分析了刚毛柽柳在不同干旱情况下脯氨酸及可溶性糖含量的不同。EST-SSR 可能是生物快速适应外界环境变化的分子装置[36]。本研究共鉴定出1 187 个多态SSR 分布在1 123 个转录本,它们可能在刚毛柽柳局部适应过程中发挥重要作用。在含有SSR 的序列中,267 个Unigenes 与公共数据库中的已知基因没有比对结果,可能代表了该物种独有的遗传资源。编码区SSR 的变异可引起基因功能的获得或丧失,5′UTRs 中SSR 的变异可影响基因的转录和翻译,3′UTR 的SSR 则影响mRNA 的剪接[37]。对刚毛柽柳含EST-SSR 基因的GO 和KEGG 功能注释发现,它们主要归类于“调节转录”、“转录因子活性”、“序列特异性DNA 结合”等GO 条目,“代谢途径”和“植物激素信号转导”等KEGG 代谢通路,一定程度反映了该植物在适应不同生境时,众多生物过程和代谢通路的基因很可能发生了较高频率的变异或表达的调节,且EST-SSRs 的变异可能在这一过程中发挥重要作用。
猜你喜欢
系统工程学报(2021年4期)2021-12-21 06:21:08
新农民(2020年15期)2020-06-22 07:38:58
新世纪智能(高一语文)(2020年12期)2020-06-01 08:14:20
河北林业科技(2020年3期)2020-03-23 13:03:16
花卉(2017年7期)2017-11-15 08:53:36
花卉(2017年7期)2017-07-20 11:10:39
中国民族医药杂志(2016年8期)2016-05-09 07:51:05
中国环境科学(2015年7期)2015-08-30 00:18:12
中国医药导报(2015年27期)2015-02-28 22:08:01
中国神经精神疾病杂志(2013年1期)2013-03-11 20:23:33
