
GNMT: a multifaceted suppressor of hepatocarcinogenesis
2021-05-11 09:04MariaSImileClaudioFeoDiegoCalvisiRosaPascaleFrancescoFeo
Hepatoma Research 2021年5期
Maria M. SImile, Claudio F. Feo, Diego F. Calvisi, Rosa M. Pascale, Francesco Feo
Department of Medical, Surgical and Experimental Sciences, University of Sassari, Sassari 07100, Italy.
Keywords: GNMT, hepatocarcinogenesis, methionine cycle
Abstract
INTRODUCTION
Accumulating evidence indicates that glycine N-methyltransferase (GNMT) plays a fundamental role in liver carcinogenesis. In GNMT-KO mice, with the deletion of exons 1-5 of theGNMTgene, HCC grows at the age 14-24 months of age, especially in female mice[1]. At the beginning of HCC growth in these mice,Wnt signaling, DNA hypomethylation, overexpression of DNA methyltransferases 1 and 3b, and activation ofGadd45a,Pak1,Mapk3, andDusp3genes of the MAPK signaling were found[1]. With the deletion of GNMT exon 1 in male mice, HCCs developed at the 8 months age[2]. Furthermore, it was found that in AflatoxinB(1)-treated GNMT-KO mice,Cyp1a2,Cyp3a44,Cyp2d22,Gsta4andAbca8agenes, involved in detoxification, were downregulated[3]. Consequently, these mice were highly susceptible to HCC induced by AflatoxinB(1). All these observations indicate that GNMT overexpression confers resistance to liver cancer development.
THE METHIONINE CYCLE
GNMT is an important component of the “methionine cycle” and, as such, plays a fundamental role in cell physiology[4,5]. In this cycle, methionine adenosyltransferases, MATI/III and MATII, convert methionine to S-adenosylmethionine (SAM). The latter has a central role in cell growth, methylation reactions, and glutathione synthesis [Figure 1]. The decarboxylation of SAM by a specific decarboxylase is implicated in the synthesis of polyamines. An end product of this synthesis, 5-methylthioadenosine, is involved in methionine re-synthesis via the “salvage pathway”[5]. Various methyltransferases including GNMT are involved in methylation reactions that transform SAM to S-adenosylhomocysteine (SAH). The latter is converted to homocysteine by S-adenosylhomocysteine hydroxylase (SAHH). Homocysteine is transformed by a specific synthetase to cystathionine, a precursor of reduced glutathione. Alternatively, homocysteine may be involved in the re-synthesis of methionine. This may be coupled to the transformation of phosphatidylcholine to phosphatidylethanolamine, operated by phosphatidylethanolamine methyltransferase (PEMT) in the Bremer pathway[6]. Phosphatidylcholine is transformed to choline, which is converted to betaine. This is joined to the synthesis of methionine from homocysteine (HCY) to methionine catalyzed by betaine homocysteine methyltransferase (BHMT). The transformation of HCY to methionine may also be associated with the folate cycle[7]. In this cycle, the methyltetrahydrofolate reductase catalyzes the conversion of tetrahydrofolate (THF) to MeTHF (5,10-methylenetetrahydrofolate), followed by the synthesis of MTHF (5-methyltetrahydrofolate), catalyzed by 5,10-methylene-tetrahydrofolate reductase, that is transformed to methionine by 5-methyltetrahydrofolate homocysteine methyltransferase[Figure 1].
GNMT catalyzes the methylation by SAM of glycine that is transformed to dimethylglycine (sarcosine). The re-synthesis of glycine catalyzed by sarcosine dehydrogenase (SARDH) is coupled to the transformation of THF into MeTHF [Figure 1]. GNMT binds to MTHF and in turn, is inhibited by MTHF[8]. Thus, GNMT induces MTHF retention, is involved in homocysteine generation, and allows MTHF-dependent remethylation of homocysteine, thus contributing to the regulation of methyl group metabolism[8-10].
The methionine and folate cycles undergo multiple interactions with cell metabolism [Figure 2]. SAM “long range interactions” involve the stimulation of GSH synthesis from HCY and the inhibition of BHMT[11-13]and MTHFR[14,15]; and therefore to methionine re-synthesis as well as of purine and deoxythymidilate synthesis. In addition, GNMT regulates the SAM/SAH ratio and SAM-dependent methyl transfer reactions.The Km value of GNMT for SAM is relatively high, and GNMT is poorly inhibited by SAH because its Ki value for SAH (35-80 μM) is higher than that for other SAM-dependent methyltransferases that are highly inhibited by SAH[8,13]. Thus, GNMT is active at SAM and SAH physiological levels (0.1-0.2 μmol/g and 0.02-0.06 μmol/of the liver, respectively). Its activity may influence SAM/SAH ratio and the activity of other methyltransferases. Besides, GNMT protein binds folate and is inhibited by MTHF[8-11,16]. The inhibition of MTHFR by SAM causes the decrease of free MTHF and is followed by the dissociation of GNMT-MTHF complex[13,16]. The rise of free GNMT avoids excessive SAM rise. On the contrary, due to the decrease in SAM concentration, MTHFR inhibition is released, MTHF availability rises, and the free GNMT falls.Therefore, by increasing the cellular folate level and thus the MTHFR-dependent homocysteine remethylation, GNMT acts as a “salvage pathway”.
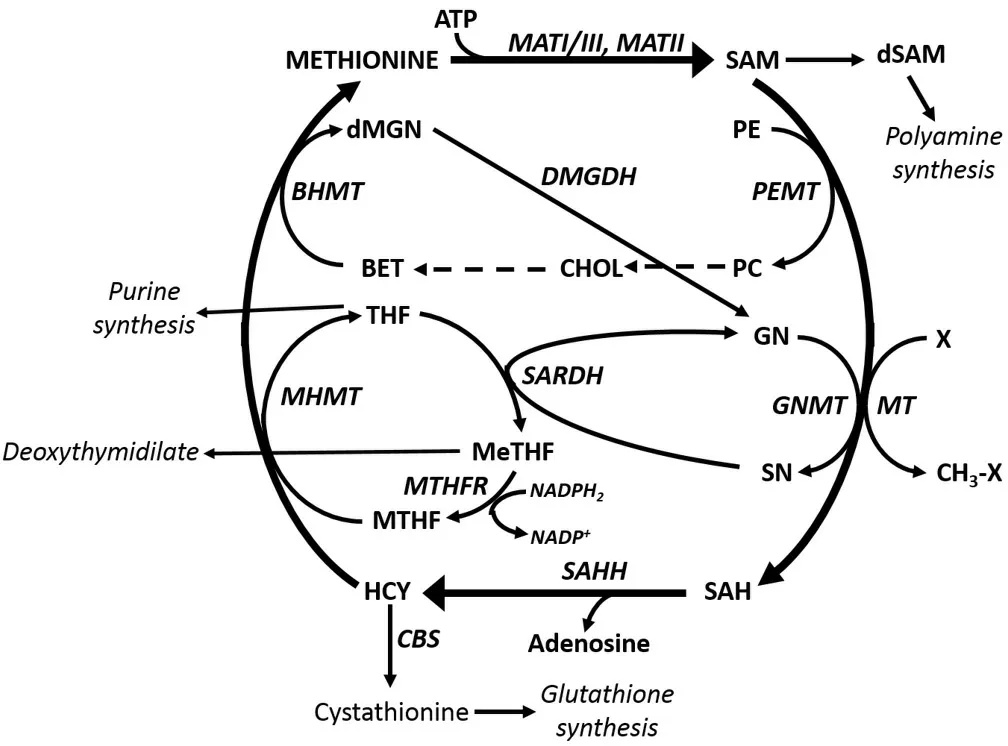
Figure 1. Methionine and folate cycles. Substrates: BET: Betaine; CHOL: choline; dMGN: di-methylglycine; dSAM: decarboxylated Sadenosylmethionine; GN: glycine; HCY: homocysteine; MeTHF: 5,10-methylenetetrahydrofolate; MHMT: methyltetrahydrofolate homocysteine methyltransferase; PC: phosphatidylcholine; PE: phosphatidylethanolamine; SAH: S-adenosylhomocysteine; SAM: Sadenosylmethionine; SN: sarcosine; THF: tetrahydrofolate. Enzymes: BHMT: Betaine homocysteine methyltransferase; CBS:cystathionine beta-synthetase; GNMT: glycine methyltransferase; MATI/III: methionine adenosyltransferase I/III; MATII: methionine adenosyltransferase II; MHMT: methyltetrahydrofolate homocysteine methyltransferase; MT: methyltransferases; MTHFR: 5-methyltetrahydrofolate reductase; ODC: ornithine decarbosylase; PEMT: phosphoethanolamine methyltransferase; SAHH: Sadenosylhomocyteine hydroxylase; SNDH: sarcosine dehydrogenase; SARDH: sarcosine dehydrogenase.
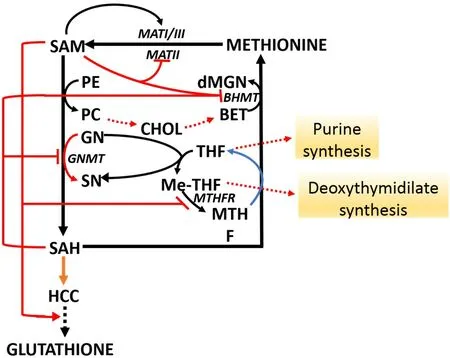
Figure 2. SAM long-range interactions. BET: betaine; BHMT: betaine homocysteine methyltransferase; CHOL: choline; dMGN:dimethylglycine; HCY: homocysteine; MATI/III: methionine adenosyltransferase I/III; MATII: methionine adenosyltransferase II;dMGN: di-methylglycine; GN: glycine; GNMT: glycine methyltransferase; MHMT: methyltetrahydrofolate homocysteine methyltransferase; Me-THF: 5,10-methylenetetrahydrofolate; PC: phosphatidylcholine; PE: phosphatidylethanolamine; SAH: Sadenosylhomocysteine; SAM: S-adenosylmethionine; SN: sarcosine. Red arrows indicate activation; red blunt arrows indicate inhibition.
THE METHIONINE CYCLE DURING LIVER INJURY
Liver injury from ethanol[17,18], acetaminophen[19], CCl4[20,21], and galactosamine[22]causes a decrease in SAM content andMAT1A:MAT2Aratio (MAT1A/MAT2A switch)[23]. In Mat1A-KO mice at 8 months[24], such decrease induces chronic SAM deficiency that is not compensated by MAT2A induction and is associated with macrovesicular steatosis in up to 50% of hepatocytes, mononuclear cell infiltration in the periportal area, and the development of HCC at 18 months. After the interruption of carcinogen administration to rats, the development of HCCs is associated with a decrease in SAM content and SAM/SAH ratio in dysplastic nodules (DN) and HCC[5]. Low SAM and SAM/SAH ratio are also present in human HCC[5]. The administration of SAM in rats and humans has been shown to decrease liver fibrosis[20,21]and prevent the development of HCCs in rats and mice[24-29].
The decrease inMAT1Aexpression in human cancer and rat liver carcinogenesis depends on CCGG sequence methylation inMAT1Agene promoter[30-33]. In contrast, the promoter ofMAT2Agene,hypermethylated and slightly expressed normal liver, is hypomethylated and transcriptionally upregulated in HCC[32,33]. The methylation ofMAT1Apromoter is also present in HepG2 and Huh7 human liver cancer cell lines. Interestingly, the treatment of HepG2 cells with the demethylating compound 5-aza-2’-deoxycytidine or with trichostatin, an histone deacetylase inhibitor, induces a rise ofMAT1Aexpression.This suggests the role of DNA methylation and histone deacetylation in MAT1A silencing[33]. In Huh7 cells,the inhibition ofMAT1Agene expression is associated with the methylation of CCGG sequence at +10 and+80 of the coding region[31]. In contrast, the overexpression ofMAT2Agene in human HCC depends on the hypomethylation of CCGG sequences in the promoter[32,33].
Interestingly, in human HCC with poorer prognosis (HCCP), higher MAT1A:MAT2A switch, CpG methylation, and histone H4 acetylation were found in human HCC with a better prognosis (HCCB)[33].Also, Mat1A/Mat2A switch, CpG hypermethylation, and deacetylation ofMat1Apromoter, associated with CpG hypomethylation and histone H4 acetylation ofMat2Apromoter occurred in HCCP of Fisher 344(F344) rats, genetically susceptible to hepatocarcinogenesis. These alterations were not found in slowly growing HCC of the genetically resistant Brown Norway (BN) rats, indicating their genetic susceptibility to liver cancer[33]. Post-transcriptional mechanisms, such as RBPs (RNA binding proteins) AUF1 and HuR are also implicated in MAT1A/MAT2A switch: AUF1 elevates mRNA decay, while HuR binding to AU-rich elements induces mRNA stabilization[34-36]. In fetal rat liver, Mat2A and HuR mRNAs are predominantly present. A sharp decrease in the HuR-MAT2A mRNA complexes and an increase in the binding of methyl-HuR to MAT2A mRNA occur during liver development. This is associated with a reduction in the levels of AUF1-MAT1A mRNA complexes, which correlate with increased MAT1A mRNA. These findings suggest that a balance between methyl-HuR, HuR, and AUF1 regulates the levels of MAT2A and MAT1A mRNAs during liver differentiation[37].
The rise of AUF1 in F344 rats and human HCCs is associated with an intense rise of MAT1A-AUF1 and MAT2A-HuR interactions[33]. miRNAs may also contribute to the MAT1A/MAT2A switch. The individual decreases of miR-664, miR-485-3p, and miR-495 in Hep3B and HepG2 cells induceMAT1Aexpression,while the overexpression of these miRNAs in Hep3B cells to induce tumorigenesis in nude mice is inhibited and increased by their knockdown[37]. Furthermore, miR-203 is shown to inhibitMAT2AandMAT2Bexpression and growth, while it increases apoptosis of HepG2 and Huh7 liver cancer cells[38].
GNMT DURING LIVER INJURY
GNMT protein and its enzymatic activity are present in the liver[39]. LowGNMTexpression was found in cirrhotic liver, HCC associated with hepatitis C virus and alcohol abuse, and in human HCC cell lines[40-43].Steatohepatitis fibrosis, cirrhosis, and HCC develop in GNMT deficient mice whereas the GNMT inducer 1,2,3,4,6-penta-O-galloyl-β-d-glucopyranoside (PGG) inhibits HCC developmentin vitroandin vivo[44].Interestingly, fast-growing HCCs are characterized by the complete absence of GNMT that may be present in low levels in slow-growing HCC. This suggests an influence of GNMT on tumor progression[45]. In pancreatic adenocarcinoma,GNMTgene is not expressed because of the methylation of its promoter[46].
AnalogousGNMThypermethylation is present in some HCC cell lines and 20% of primary HCCs[47].Interestingly, GNMT binds some carcinogenic compounds, including aromatic hydrocarbons and aflatoxins. This prevents the formation of DNA adducts and carcinogen cytotoxicity[48-50]. Studies of GNMT,performed in HCC of Taiwanese men, indicated thatGNMTmay be a tumor susceptibility gene for this tumor[51]. Furthermore, the presence of GNMT was not detected in SK-Hep1, Hep 3B, Huh-7, and HA22T human HCC cell lines. The investigation of a Taiwanese liver cDNA library allowed the isolation of the clone-9-1-2 full-lengthGNMTcDNA. This clone, compared with the GNMT cDNA sequence, exhibited 4 nucleotide differences, causing the change of an amino-acid. Immunohistochemical staining with the rabbit anti-recombinant GNMT serum showed that GNMT protein almost completely disappeared in cancer cells[51]. Other researches[52,53]revealed the GNMT short tandem repeat polymorphism (STRP1) and single nucleotide polymorphism were not associated with a rise in the risk of prostate cancer in Taiwanese men of European descendants[53]. In prostate cancer, it was identified as an androgen-responsive element in the first exon of theGNMTgene, binding androgen receptorin vitroandin vivo[54,55]. This indicates that androgen is an important regulator ofGNMTgene. Furthermore, GNMT-KO mice, lacking exons 1 of theGNMTgene,develop HCC. This was associated with hypermethioninemia and elevated levels of serum aminotransferase and liver SAM[55,56]. In about 60% of these mice, high glycogen storage in the liver was detected and HCC prevalently developed in female mice at 14-24 months of age[56].
An important contribution to understanding the role of GNMT liver carcinogenesis was the demonstration in GNMT-KO mice of 7- and 35-fold rises in free methionine and SAM, respectively, associated with a 3-fold decrease of SAH level[57]. At the early stages of HCC development, global DNA hypomethylation and aberrant expression of DNA methyltransferases 1 and 3b were observed[1]. In a male GNMT-KO model with disrupted GNMT exon 1, high serum aminotransferases, methionine, and SAM levels were associated with liver steatosis, fibrosis, and HCC development at the age of 8 months[2]. Tumors in these mice were characterized by the activation of Ras and Jak/Stat (Janus kinase/signal transducer and activator of transcription) pathways, and suppression of Ras inhibitors RASSF 1 and 4, Jak/Stat inhibitors SOCS 1-3,and cytokine-inducible SH2-protein[2]. Hypermethylation of Rassf1 and Socs2 promoters and binding of trimethylated lysine 27 of histone 3 to these 2 genes occurred in HCCs from GNMT-KO mice[2]. This indicates that the presence of aberrant methylation of DNA and histones caused the epigenetic modulation of carcinogenic pathways. Some interactions also occur between GNMT and LKB1/AMPK signaling;indeed, HCC cells from GNMT-KO mice lacking exon 1 showed, like human HCCs, overexpression of theRasgene that mediated the hyper-activation of Lkb1 with consequent AMPK activation[3]. This requires Erk2 and p90rsk activity and Rasgrp3 expression. Interestingly, in HCCP, reduced levels of GNMT,phosphorylation of AMPKα at Thr172, and overexpression ofRAS,LKB1, andRASGRP3have been reported[57].
GNMT EXPRESSION IN TRANSCRIPTOMIC CLASSIFICATIONS OF HCC
Human HCCs have been classified based on metabolic features[58]in tumors with better (HCCB) and poorer(HCCP) prognosis[59]. Interestingly, in HCCP, a higher MAT1A:MAT2A switch, CpG methylation, and histone H4 acetylation occur, with respect to HCCB[33]. Analogous alterations were found in HCC of F344 rats, genetically susceptible to hepatocarcinogenesis. In contrast, these alterations were absent in slowly growing HCC of the genetically resistant BN rats[33]. Interestingly, it was observed that genes involved in methionine metabolism, such asBHMTandGNMT, are also involved in the resistance to liver cancer[59]. A gene expression signature with the highest expression of proliferation-relatedCTGF,c-MYC, andPCNAgenes, was associated with the lowest expression of the onco-suppressorsBHMT,DMBT1,DUSP1,GADD45gandGNMTgenes, in HCCP[59]. An overexpression of the onco-suppressorsBhmt,Dmbt1,Dusp1,Gadd45g,GNMT,Napsa,Pp2ca, andPtpn13was found in HCCs of BNA rats[59,60]. In light of these findings,it is interesting that genes involved in methionine metabolism, such asBHMTandGNMT, are also involved in the resistance to liver cancer[59]. The mechanisms by which GNMT participates in tumor prevention/suppression in humans are not completely clear. It has been shown[1]that GNMT supports methylene-folate dependent pyrimidine synthesis and formyl-folate dependent purine synthesis [Figure 2].It also minimizes uracil incorporation into DNA because of folate depletion and translocates into the nucleus during prolonged folate depletion. According to these findings, loss ofGNMTimpairs nucleotide biosynthesis, whereasGNMToverexpression enhances nucleotide biosynthesis and improves DNA integrity by reducing uracil misincorporation in DNA bothin vitroandin vivo[1].
GNMT AND LIVER CANCER SUPPRESSION
Studies on GNMT-KO mice indicate thatGNMTis a suppressor gene and confirm previous findings suggesting that down-regulation ofGNMTgene expression may be involved in the pathogenesis of liver cancer. Low levels ofGNMTactivity were detected in fetal rabbit liver about 20 days post-fertilization, it increased to higher levels after birth, reaching maximum in the adult liver[45]. In fast-growing rat HCCs,GNMT activity is absent, but it can be detected in the slower-growing HCCs at lower levels than in normal adult rat liver, suggesting a role of GNMT in HCC progression[42]. Furthermore, GNMT could be involved in stress responses induced by low-dose irradiation of mouse liver[59]. GNMT also regulates genes implicated in the detoxification and anti-oxidation pathways, and Benz(a)pyrene carcinogenicity[60]. It is also an essential regulator of Complex II activity in the mitochondrial electron transport chain[61].
GNMT-KO has allowed the study of liver progenitor cells and their role in hepatocarcinogenesis. E. Farber first hypothesized that progenitor cells resistant to liver injury proliferate to form preneoplastic dysplastic nodules and HCCs by the “resistant hepatocyte protocol”[62]. The GNMT-KO mice model allows a detailed study of these cells. In these mice livers, hyperplasia of progenitor OV-6 positive cells occurs at 8 months, a process that increases at 18 months when HCC develops and expresses Fat10, the marker for progenitor liver cells[62]. In an analogous liver cancer model, in mice that are fed diethyl-1,4-dihydro-2,4,6-trimethyl-3,5-pyridine decarboxylate (DDC), stem cell/progenitor cell (SCP) are upregulated as indicated by microarrays, and are prevented by SAM[63].
The mechanisms of tumor suppression mediated by GNMT are complex and probably not completely known. The protein mTOR (mechanistic target of rapamycin) controls cell growth and translation by the assembly of multi-protein signaling complexes, the best described of which is mTORC1 (mechanistic target of rapamycin complex 1)[64]. In this complex, mTOR is associated with RAPTOR (regulatory associated protein of mTOR), LST8, and PRAS40 proteins[65]. The increase in mTORC1 activates S6K1/2 proteins and represses 4E-BPs[65]. RAPTOR over-expression activates AKT via the inhibition of the negative feedback loop from S6K1 to ISR (insulin receptor substrate)[65]. The RAPTOR/mTOR complex, overexpressed in HCC, interacts with GNMT[65][Figure 3]. A decrease in mTORC1 signaling, induced byGNMToverexpression, inhibits G2/M cell cycle progression, and Huh-7 cell proliferation[65].
PREX2 (phosphatidylinositol 3,4,5-trisphosphatedependent Rac exchanger 2) regulates the small guanosine triphosphatase, Rac, and induces the growth and migration of HCC cells by the PTEN-AKT signaling.Moreover,PREX2overexpression in HCC inhibits the activity of PTEN (phosphatase and tensin homolog),which regulates HCC cell invasiveness[66]. The inactivation of PTEN by PREX2, in turn, inactivates AKT.The opposite occurs by the knockdown ofPREX2expression[66]. The inhibition of AKT signaling is also induced by GNMT interaction with the PTEN inhibitor, PREX2[67,68]. This interaction enhances the proteasomal degradation of PREX2 by the E3 ubiquitin ligase, HectH9 [Figure 4]. Prex2 protein overexpression, in association with Akt activation, occurs in the liver of GNMT-KO mice[66]. Elevated levels of PREX2 were also found in human HCC and associated with decreased survival. In these HCCs,PREX2 mRNAexpression is not decreased, indicating that a post-translational alteration ofPREX2expression occurs[67].
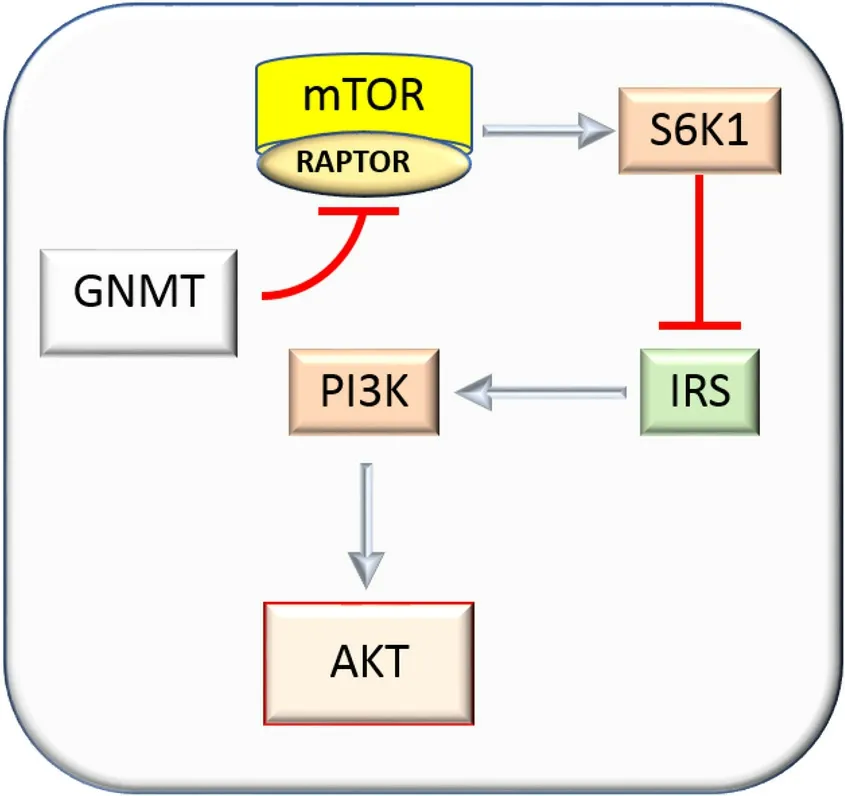
Figure 3. Mechanisms of inhibition of hepatocellular carcinoma growth by glycine methyltransferase. The inhibition of the RAPTOR/mTOR complex, impedes S6K1 activation and the feedback negative effect of S6K1 on the PI3K/AKT axis mediated by IRS.GNMT: glycine N-methyltransferase; mTOR: mechanistic target of rapamycin; IRS: insulin response element; PI3K: phosphoinositide-3-kinase.

Figure 4. Inhibition of PREX2proteasomal degradation by GNMT. AKT: Murine thymoma viral oncogene homolog; GNMT: glycine Nmethyltransferase; HectH9: E3 ubiquitin ligase; PI3K: phosphoinositide-3-kinase; PIP2: phosphorylating phosphatidylinositol-4,5-bisphosphatde; PIP3: phosphatidylinositol-3,4,5-triphosphate; PREX2: phosphatidylinositol 3,4,5-trisphosphatedependent Rac exchanger 2.
The above mechanisms of inhibition of the suppressive effects of GNMT are epigenetic. However, the transport of rat GNMT into liver nuclei, where it interacts with chromatin, has also been observed[68,69].Nuclear GNMT has different and sometimes contradictory effects. The GNMT nuclear localization induces apoptosis, independently of its catalytic activity or folate binding capacity[70]. The anti-proliferative effect of GNMT is partially reversed by the pan-caspase inhibitor, zVAD-fmk[69].
GNMT translocation into the nucleus supports pyrimidine and purine synthesis, and lessens uracil incorporation into DNA in cells with folate depletion[70,71]. Its loss impairs nucleotide biosynthesis[72]. GNMT suppressor activity also relates to the regulation of cytochromeP450-1A1gene expression[71,72]. The polymorphism of this gene increases the susceptibility to breast[73], cervical[73], and lung[74]cancers. The tobacco-derived polycyclic aromatics hydrocarbons are implicated in the risk of HCC in chronic HBV carriers, andCYP1A1polymorphism is an important modulator of the hepatocarcinogenic effect of these carcinogens[75].
Recent research has shown that NRF2 overexpression is implicated in HCC development[76]. NRF2 translocation to the nucleus induces the dimerization with sMAF proteins[77]. The complex NRF2/sMAF binds antioxidant response element (ARE), activating the transcription of target genes [Figure 5]. The transcription factor, NRF2, is a main regulator of the antioxidative and detoxification responses and propels the progression of cancer, the formation of metastases, and the resistance to therapy[78]. Interestingly,nuclear GNMT interacts with the promoter region of the genes encoding NRF2[79].
In the cirrhotic liver of cholestatic patients and animal models of liver cholestasis and cirrhosis, miR-873-5p is inversely correlated withGNMTexpression; high circulating miR-873-5 is present in cholestatic patients[80]. The treatment with anti-miR-873-5p in Mdr2-/-mice with bile duct ligation recovers GNMT levels and improves the inflammation and fibrosis by counteracting hepatocyte apoptosis and cholangiocyte proliferation[80]. Furthermore, in patients with non-alcoholic fatty liver disease (NAFLD) and a preclinical murine non-alcoholic steatohepatitis (NASH) model, miR-873-5p controlsGNMTexpression of hepatocytes, leading to the disruption of mitochondrial functionality[81]. GNMT is an essential regulator of Complex II activity in the electron transport chain of mitochondria[81]. The upregulation of miR-873-5p in the liver of NAFLD/NASH patients is correlated with GNMT depletion. Importantly, NASH therapies based on anti-miR-873-5p, sort out lipid accumulation, inflammation, and fibrosis by enhancing fatty acid β-oxidation in mitochondria[81]. GNMT participates in the regulation of metabolic pathways and mitochondrial functionality through the regulation of Complex II activity in the electron transport chain[81].
The observations that GNMT knockout mice developed HCC and GNMT prevented aflatoxin-induced carcinogenicity and inhibited HCC cell proliferation, motivated the research on GNMT inducers for HCC therapy. The extract of the active component of Paeonia lactiflora Pall, PGG, was identified as a GNMT inducer. PGG treatment sensitized Huh7 cells to sorafenib treatment[82]. These findings explain why PGG could have therapeutic potential for the treatment of HCC[44]. However, at present, cancer therapy that stems from alterations in the methionine cycle is based on the inhibitory effects of SAM. Different studies have documented the decrease of hepatic SAM content during acute and chronic alcoholism[17,83], and the prevention of fatty liver and GSH decrease by the administration of exogenous SAM[17]. Na+, K+-ATPase is an integral plasma membrane enzyme that plays a key role in the physiology and structure of hepatocytes,where it maintains the electrochemical gradients for Na+and K+across the cell membrane. Its activity is inhibited by ethanol that determines a decrease in GSH liver content and steatosis. SAM accelerates the recovery of these parameters after ethanol withdrawal and also protects (Na+, K+) ATPase activity and GSH content of isolated hepatocytes from the deleterious effect of ethanol[17,84,85]. The maintenance of a high GSH level by SAM determines its beneficial effect that indeed is prevented by 1-chloro-2,4-dinitro-benzene, a compound that depletes cell GSH[86].

Figure 5. Effects of Nrf2 nuclear translocation. After nuclear translocation, NRF2 dimerizes with sMAF proteins, and together they bind ARE to activate the transcription of NRF2 target genes. Some processes regulated by NRF2 target genes are shown. ARE: Antioxidant response element.
Present knowledge indicates that both epigenetic and genetic mechanisms may be involved in cancer suppression by GNMT. The epigenetic mechanisms implicate the inhibitory effects of GNMT on AKT signaling, which in turn, is linked to its interaction with MTOR and PREX2[64-67]. Less clear are the genetic mechanisms responsible for the suppressor activity of GNMT. A connection of GNMT with the regulation ofCyP1A1andNRF2genes is suggested[71-76], bsut further work is necessary to clarify this aspect of theGNMTsuppressor activity.
DECLARATIONS
Authors’ contributions
Organized and wrote the review: Simile MM, Feo CF, Calvisi DF, Pascale RM, Feo F All Authors contributed equally.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by Grant: Fondazione di Sardegna: R.M. Pascale, 2017.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.

- Hepatoma Research的其它文章
- Prevention of hepatitis B virus recurrence
- Extrahepatic cancer risk after liver transplantation for hepatocellular carcinoma: incidence, risk and prevention
- Systemic therapies for hepatocellular carcinoma: an evolving landscape
- Gut microbiome profiles associated with steatosis severity in metabolic associated fatty liver disease
- Steatohepatitic hepatocellular carcinoma
- Epidemiology and aetiology of hepatocellular carcinoma in Sub-Saharan Africa