
团簇Ni4P的催化性质和磁性
2021-05-07 10:38:10吕孟娜方志刚井润田王智瑶侯欠欠
辽宁科技大学学报 2021年1期
吕孟娜,方志刚,秦 渝,井润田,王智瑶,侯欠欠,许 友,陈 林
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
非晶态合金的结构不同于晶态金属,其原子排列具有长程无序的特点,独特的结构使其具有许多常规晶体所不敌的优异性能[1],在众多领域具有潜在的应用前景。其中,Ni-P非晶态合金已成为近年来发展较为快速的一种新型催化材料[2],该合金表面高度不饱和,且原子配位数少,因而具有较强的催化活性,常作为催化剂用于电极催化[3]、加氢脱氢[4]和异构化等反应。此外,优异的磁学性能[5-7]使其成为软磁材料的首选,由于非晶态合金中没有阻碍磁畸壁移动晶界、亚晶界及第二相颗粒,所以很容易磁化且磁致损失小,能够提高能源利用率。
目前对Ni-P非晶态合金体系宏观方面的研究较多,而微观方面的理论探究较少。Jiang等[8]通过电化学极化测试,测量了该镀层的电磁干扰屏蔽效能,并对其耐蚀性进行了表征,发现Ni-P可提高Ni-P/Cu-Ni镀层的耐蚀性,且大大提高了镀层的电磁屏蔽性能,随着Ni-P沉积时间的延长,Ni-P/Cu-Ni镀层的EMI-SE和耐蚀性也随之提高。Dhanapal等[9]采用脉冲电沉积法合成两种不同占空比的Ni-P合金,证明Ni-P合金的矫顽力和饱和磁化强度随外加电流、占空比和磷量的变化而变化,且对于相同的外加电流,占空比的增加会降低矫顽磁力和顽磁力值。Xie等[10]采用化学沉积法制备了纳米碳纤维负载的非晶态Ni-P合金催化剂,以硝基苯液相加氢为探针考察其催化活性,发现具有独特的鳞片状结构和较大的比表面积,具有较高的催化活性。本文以团簇Ni4P作为Ni-P体系的局域结构,从微观角度探究其催化性质和磁学性质,期望能为该体系宏观及微观角度的研究提供有价值的理论依据。
1 优化构型与催化性质
1.1 优化构型
依据拓扑学原理[11],设计出团簇Ni4P可能存在的10种初始构型。运用密度泛函理论[12],对设计的初始构型于B3LYP/Lanl2dz水平下进行优化,排除虚频构型和稳定性略差的相同构型后,得到6种优化构型。对过渡金属镍采用Hay等[13]的18-eECP的双ξ基组(3s、3p、3d/2s、2p、2d),并对类金属磷加极化函数ξp.d=0.55[14]。所有计算均使用Gaussian09程序在启天M7150微机上完成。
采用上述方法得到的优化构型如图1所示,二、四重态下的稳定构型各为3种。将能量最低的稳定构型1(4)作为基准,设其能量为0 kJ/mol,其余构型由相对能量从低到高排列编号,各构型右上角括号内的数字代表该构型的重态。构型稳定性与其能量相关,能量越低,稳定性越好。团簇Ni4P的稳定性强弱关系为1(4)>1(2)>2(4)>2(2)>3(4)=3(2)。

图1 团簇Ni4P的优化构型图Fig.1 Optimized configurationsof cluster Ni4P
1.2 催化性质
在前线轨道理论中,HOMO和LUMO分别为最高占据分子轨道和最低未占分子轨道。分子中存在着类似于单个原子的“价电子”(前线电子),当分子间发生化学反应时,前线轨道起关键性作用。分析各原子对HOMO、LUMO轨道贡献情况可探究团簇Ni4P的催化活性位点,各构型的HOMO和LUMO贡献率数据如表1所示。
ΣNi和ΣP分别代表Ni、P原子对HOMO轨道和LUMO轨道的总贡献率。除构型3(4)和3(2)的Ni、P原子对HOMO、LUMO轨道贡献率完全相同外,其余构型的Ni、P原子对HOMO、LUMO轨道贡献率数值均不相同且均大于零,说明Ni、P原子对各构型的催化活性均起作用,但催化能力均不同。构型2(2)的Ni原子对HOMO轨道的贡献率最大,P原子对HOMO轨道的贡献率最小,证明该构型的Ni原子是HOMO轨道的主要贡献者。重态不同但空间结构相同的构型1(4)和1(2)的Ni、P原子对HOMO、LUMO轨道的贡献率相差极小,3(4)和3(2)的Ni、P原子对HOMO、LUMO轨道的贡献率完全相同,说明Ni、P原子对轨道贡献率受空间结构的影响。四重态构型中Ni和P原子对HOMO、LUMO轨道的贡献率与相对应的二重态构型相差较小,说明自旋多重度对轨道贡献率影响较小。从整体分析,团簇Ni4P的Ni原子对HOMO、LUMO轨道的平均贡献率均大于P原子,说明Ni原子是前线轨道的主要贡献者,催化活性主要由Ni原子提供,Ni原子最有可能成为团簇Ni4P的催化活性位点。相比于Ni原子,P原子对HOMO、LUMO轨道的平均贡献率虽然较低,但几乎占总贡献的1/5,所以P原子对团簇Ni4P催化活性的影响亦不可忽略。说明团簇Ni4P的催化活性是以Ni原子为主,P原子为辅所提供的。
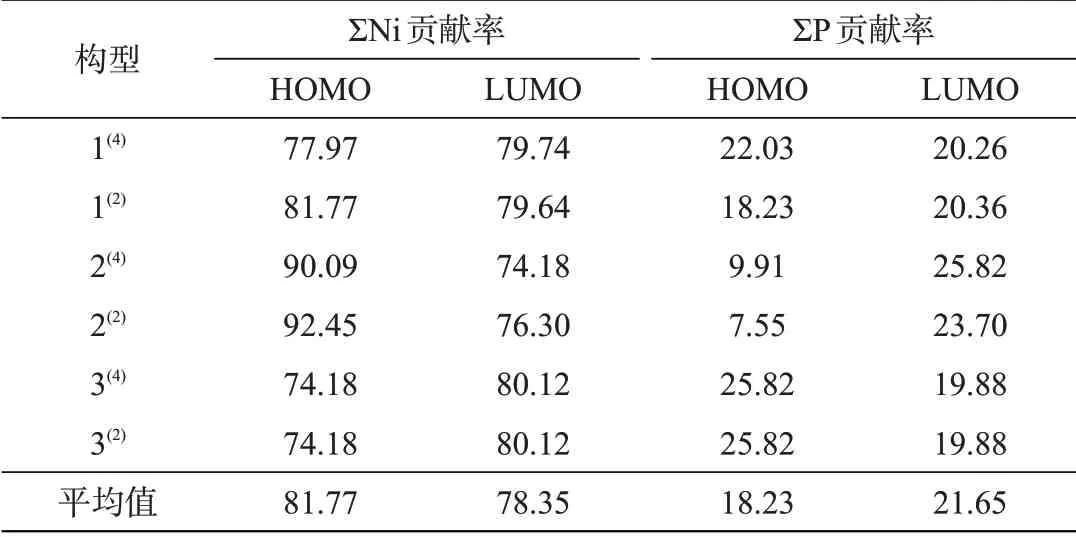
表1团簇Ni4P各原子HOMO、LUMO贡献率,%Tab.1 Contribution ratesof HOMOand LUMOin Ni4Pcluster,%
为了更加直观地分析团簇Ni4P的催化性质,根据表1中Ni、P原子对HOMO、LOMO轨道贡献率绘制折线图如图2所示。团簇Ni4P各构型的Ni原子对HOMO、LUMO轨道贡献率均大于P原子,说明Ni原子对其催化活性起主要贡献。各构型的Ni原子对HOMO、LUMO轨道贡献率变化趋势均与P原子完全相反,说明两原子间具有拮抗作用。此外,构型2(4)~2(2)的Ni原子内部和P原子内部HOMO、LUMO轨道贡献率变化趋势相同,其余构型间原子内部贡献率变化趋势均相反,说明构型2(4)~2(2)的Ni、P原子内部存在协同作用,而其余构型为拮抗作用,则构型2(4)~2(2)既具有拮抗作用又存在协同作用,二者相互制约使其表现出更好的催化活性。
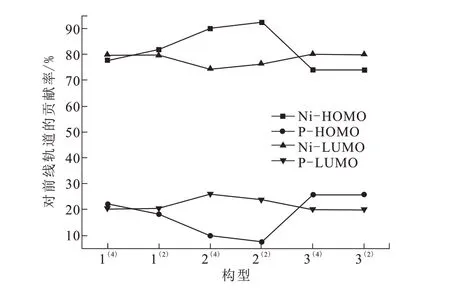
图2 团簇Ni4P各构型的Ni、P原子对前线轨道的贡献Fig2 Contributionsto frontier orbitalsby Niand Patomsin Ni4Pcluster configurations
2 磁学性质
物质的磁性与组成物质的原子微观结构有关。原子核外存在着自旋向上且数值为正的α电子和自旋向下且数值为负的β电子,当二者的自旋电子数不相等时,即代表有未成对电子数,这时物质具有永久磁矩。因此,可以通过分析团簇Ni4P各构型的s、p、d轨道未成对电子数来探究其磁学性质,将各轨道未成对电子数列于表2。
s轨道未成对电子数波动范围较小,为-0.175 55~0.187 51,p轨道的未成对电子数波动范围相对较大,为-0.268 95~0.570 37,而d轨道未成对电子波动范围最大,为0.241 76~3.268 64。除构型2(4)外,s轨道未成对电子数均小于p轨道,是因为s壳层易被成对电子占据。其中,构型1(4)~2(2)既有自旋向上的α电子又有自旋向下的β电子,d轨道未成对电子数远大于s、p轨道,且s、d轨道四重态未成对电子数均大于二重态,p轨道四重态未成对电子数略低于二重态,说明这四种构型的磁性主要是由d轨道上存在的自旋向上的α电子贡献,且四重态贡献更为显著。构型3(4)~3(2)的各轨道未成对电子数完全相同且均为正值,则二者的磁性由自旋向上的α电子提供,又因p轨道未成对电子数大于s、d轨道,说明两者的磁性主要由p轨道贡献。四重态构型1(4)和2(4)的未成对电子数为3,二重态构型未成对电子数为1,这符合自旋多重度[15]的定义,但四重态构型3(4)的未成对电子数与二重态构型3(2)完全一致,都为1,且两者的能量也完全相同,可能是因为这两构型之间存在动态平衡,在过渡态转化时极可能发生构型3(4)→3(2)的转化,可通过研究该构型异构化反应对其进行理论验证。
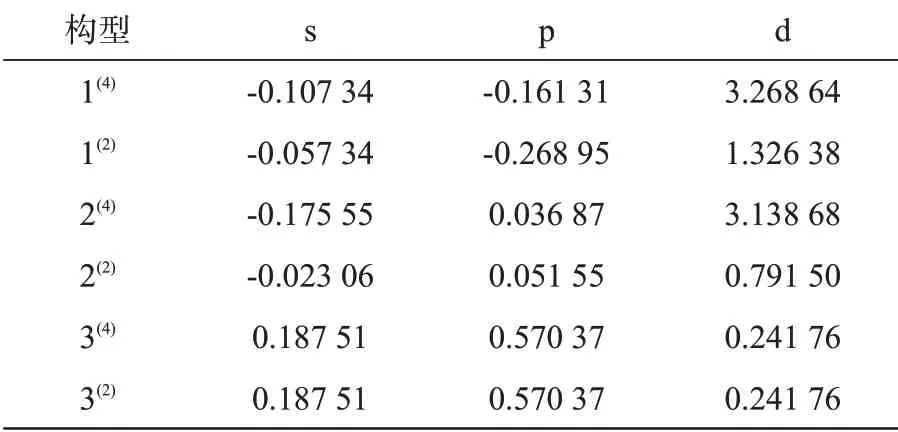
表2 团簇Ni4P各构型中s、p、d轨道未成对电子数Tab.2 Number of unpaired electrons on s、p、d orbitals in various Ni4Pcluster configurations
原子总磁矩由电子轨道运动产生的电子轨道磁矩和电子自旋产生的自旋磁矩构成。磁矩(μs)等于自旋向上的电子占据数减去自旋向下的电子占据数,单位为玻尔磁子μB。
图3为团簇Ni4P各构型Ni、P原子总磁矩变化趋势图。磁矩为正值对各构型磁性有利,反之不利。各构型的Ni原子磁矩均大于零,且大于P原子,证明Ni原子对团簇Ni4P的磁性起主要作用。构型1(4)~2(4)的Ni、P原子均随能量的增加磁矩先减小后增加,且Ni原子均大于零,而P原子均小于零,说明这三个构型之间具有协同作用,Ni原子对磁性有利,而P原子对磁性不利。构型2(2)~3(2)的Ni原子随能量的增加磁矩减小且均大于零,而P原子随能量的增加磁矩增大也均大于零,说明这三个构型之间具有拮抗作用,但均对磁性有利。构型2(4)~3(2)随能量的增加Ni、P原子磁矩绝对值逐渐接近,说明能量的变化在一定程度上使其磁学性质接近;构型1(4)和2(4)的Ni、P原子磁矩之和相等且大于其余构型,证明构型1(4)和2(4)的磁性强度相当且强于其他构型;此外,构型1(4)的Ni、P原子的磁矩绝对值均大于构型2(4),说明构型1(4)的磁矩分布范围最大,构型2(4)的磁矩分布次之,即构型1(4)的磁性略强于构型2(4)。
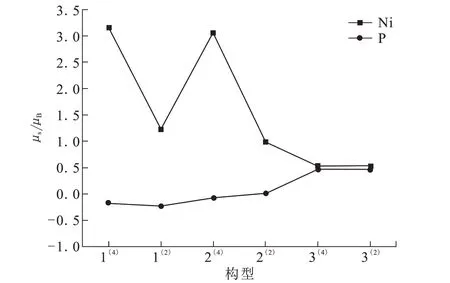
图3 团簇Ni4P各构型Ni、P原子磁矩Fig.3 Magnetic momentsof Niand Patomsin various Ni4P cluster configurations
3 自旋态密度
为了更加深入地探究团簇Ni4P各构型的s、p、d轨道对磁性的贡献情况,绘制s、p、d轨道上的电子在不同能量范围的分布情况,即电子自旋态密度图,如图4所示。各轨道的自旋向上和自旋向下的态密度曲线对能量积分之和为对应轨道的成单电子数。
构型1(4)~2(2)中,d轨道中最高峰A和A'处的峰值相差较大,峰B和B'处的态密度分布明显不对称。构型1(4)和2(4)不仅在B和B'处的不对称性明显大于构型1(2)和2(2),而且两构型的态密度曲线存在错位现象。构型1(4)和1(2)在C和C'处的态密度分布也呈现些微不对称现象。这些说明d轨道对这四个构型磁性贡献大小关系为:1(4)>2(4)>1(2)>2(2)。构型1(4)~2(2)的s、p轨道的态密度分布几乎完全对称,且p、d轨道的态密度分布明显大于s轨道,说明d轨道对这四个构型磁性起主要贡献作用,p轨道次之,s轨道贡献最弱。构型3(4)~3(2)的s轨道在D和D'处的态密度分布明显不对称,E和E'处为电子自旋方向改变后生成的峰且峰值具有差异,尽管s轨道的峰值较p、d轨道较小,但该轨道对两构型磁性产生的影响仍不可忽略。构型3(4)~3(2)的p、d轨道都存在错位现象,p轨道中F和F'处和d轨道中B和B'处的态密度分布均存在着微弱的不对称现象,且d轨道中最高峰A和A'处的峰值虽相差较小但亦不能忽略,说明s、p、d轨道对两构型的磁性均具有一定的贡献作用。
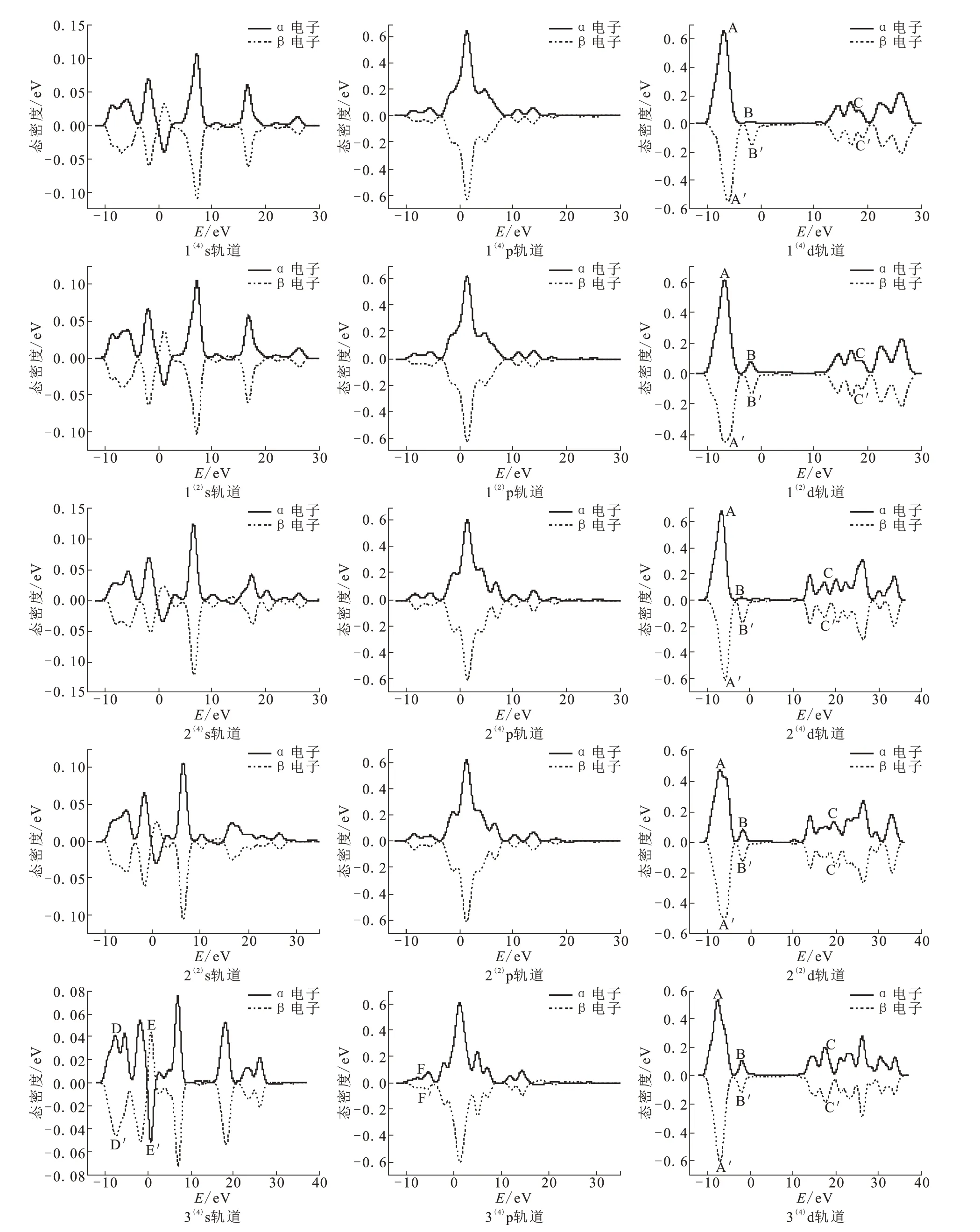
图4 团簇Ni4P各稳定构型的s、p、d轨道态密度分布Fig.4 Distributionsof statedensity of s、p、d orbitalsin stablecluster Ni4Pconfigurations
各构型的s轨道上0~5 eV和10~15 eV处均出现明显的α、β电子自旋方向改变,构型3(4)和3(2)的p轨道上15~20 eV处也出现较为微弱的α、β电子自旋方向改变的情况,是因为电子自旋具有不确定性,导致其α、β电子发生部分偏转。
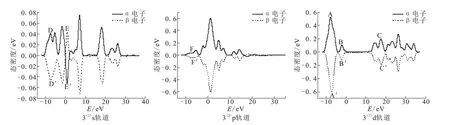
续图4 团簇Ni4P各稳定构型的s、p、d轨道态密度分布Fig.4 Distributionsof statedensity of s、p、d orbitalsin stablecluster Ni4Pconfigurations
4 结论
(1)团簇Ni4P各优化构型中,Ni、P原子对轨道贡献率受空间结构的影响,但受自旋多重度的影响极小;Ni原子是前线轨道的主要贡献者,P原子对其贡献亦不可忽略,则催化活性是以Ni原子为主,P原子为辅,Ni原子最可能为团簇Ni4P的催化活性位点;构型2(4)~2(2)的Ni、P原子之间具有拮抗作用,又存有协同作用,二者相互制约使其表现出更好的催化活性。
(2)构型1(4)~2(2)的磁性由d轨道上存在的自旋向上的α电子贡献,且四重态贡献更为显著;3(4)~3(2)的磁性主要由p轨道贡献;Ni原子对团簇Ni4P的磁性起主要贡献作用,且构型1(4)的磁矩分布范围最大,构型2(4)的磁矩分布次之;电子自旋具有不确定性,导致其α、β电子发生部分偏转。
猜你喜欢
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
深圳大学学报(理工版)(2015年6期)2015-11-26 12:33:48
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
航天返回与遥感(2014年4期)2014-07-31 17:47:47
无机化学学报(2014年4期)2014-02-28 17:31:23
无机化学学报(2014年4期)2014-02-28 17:31:08
应用技术学报(2014年1期)2014-02-28 14:52:11
河南科技(2014年23期)2014-02-27 14:18:52
