
基于PCR熔解曲线技术的结核分枝杆菌Spoligotyping基因分型的临床应用研究
2021-05-06 02:02蓝如束覃慧芳黄莉雯苏华斌罗淋尹
中国人兽共患病学报 2021年4期
蓝如束,叶 婧,罗 丹,覃慧芳,黄莉雯,苏华斌,罗淋尹,林 玫
结核病仍然是严重危害人类健康的传染病之一,每年大约有1 040万人感染,140万人死于结核,形势十分严峻[1]。近年来,结核分枝杆菌(Mycobacteriumtuberculosis,MTB)基因分型方法普遍用于结核病疫情处置和结核病分子流行病学研究。目前,用于MTB基因分型的主要方法有限制性片段长度多态性(IS6110-RFLP)、间隔区寡核苷酸分型技术(Spoligotyping)、可变串联重复序列技术(VNTR)等[2]。这些方法普遍操作繁琐、耗时长、结果判读主观性大,操作人员需要经过严格培训。其中间隔区寡核苷酸分型技术(Spoligotyping)方法是最常用基因分型方法之一,传统检测技术是利用PCR方法扩增间隔序列,扩增产物与固定在杂交膜上的 43 个合成的间隔序列捕获探针杂交,根据显影结果判读相应的间隔序列是否存在,进而得到菌株的基因型特征。其操作步骤多且非常繁琐,结果需要人工判读。近两年来,厦门致善公司研制出一种利用PCR熔解曲线技术对结核分枝杆菌进行spoligotyping基因分型,该技术仅需3 h完成检测,实现结果自动化判读。本研究旨在应用Mcspoligotyping方法对临床样本进行检测,评价其在结核病疫情处置及结核病分子流行病学研究中的应用价值。
1 材料和方法
1.1标本来源 949株菌株来源于2017年广西结核病耐药监测,监测期间收集临床病人痰标本,应用罗氏培养基传统固体分离培养方法获得阳性培养物,经硝基苯甲酸(PNB)和噻吩-2-羧酸肼(TCH)鉴别培养基确定为MTB。质控标准菌株H37Rv由中国疾病预防控制中心国家结核病参比实验室提供。
1.2仪器和试剂 ZEESAN SLAN 96实时荧光定量PCR仪(厦门致善生物科技股份有限公司)、细菌超声分散计数仪(广东体必康生物有限公司)。结核分枝杆菌McSpoligotyping分型检测试剂盒购买于厦门致善生物科技股份有限公司,试剂包含PCR MixA、PCR MixB、PCR MixC、MTBC酶、阴性和阳性对照。分枝杆菌菌种鉴定培养基购自珠海贝索生物技术有限公司。
1.3 方 法
1.3.1DNA提取 采用CTAB法进行提取DNA[3],在1.5 mL离心管加入400 μL 1×TE缓冲液并取满环生长良好结核分枝杆菌,置于80 ℃水浴30 min灭活,将灭活好菌液在细菌超声分散计数仪超声1 min,使细菌充分分散,加入溶菌酶使细菌充分破壁,然后用SDS/蛋白酶K混合,用CTAB/NaCl溶液沉淀非核酸细胞碎片,用氯仿/异戊醇提取DNA,在提取液中加入异丙醇使DNA沉淀,沉淀物经洗涤且自然干燥后,加入50 μL 1×TE缓冲液使沉淀物充分溶解,置-20 ℃保存备用,作为基因分型和WGS检测的DNA 模板。
1.3.2McSpoligotyping检测方法
1.3.2.1McSpoligotyping方法原理 是采用荧光PCR熔解曲线法,选择结核分枝杆菌所携带的43个特定的间隔序列作为检测模型。首先,在高度同源的同向重复序列(DR,direct repeat)上设计一对通用引物,并利用这对通用引物扩增出间隔序列(S1-S43);其次,根据每个间隔序列的DNA序列设计能够与之特异杂交的荧光探针。每条荧光探针都带有特征性的荧光标记和熔点温度组合。当PCR产物中存在某一间隔序列时,相应的荧光探针就能与之杂交形成特征的熔解曲线峰;最后,通过这些熔解曲线峰的组成确认待检样品中所存在的间隔区,从而推断样本中所携带的结核分枝杆菌型别。对于每份检测样本需3个反应对其进行检测,对应扩增试剂中的PCR MixA、PCR MixB、PCR MixC。
1.3.2.2PCR反应及熔解曲线分析 所有试剂从冰箱取出平衡至室温。PCR反应液配液标准为:取n×19.75 μL PCR Mix(A/B/C)(n根据反应管数确定)和n×0.25 μL MTBC酶混合液加入到1.5 mL离心管中,振荡混匀数秒,然后瞬时离心,配成PCR反应液,以每管20 μL分装于PCR 反应管。用微量加样器向每个PCR反应管加入相应的DNA样品,并设阴阳性对照。采用致善SLAN96实时PCR仪进行扩增及熔解曲线分析。反应体系设为25 μL,PCR程序设置如下:50 ℃ 5 min→95 ℃ 10 min→(95 ℃ 15 s→57 ℃ 15 s→72 ℃ 15 s)×50个循环。熔解曲线分析程序设置如下:95 ℃ 1 min→35 ℃ 1 min→35 ℃~90 ℃ 以 0.04 ℃/s的升温速率进行熔解分析,且在此阶段采集FAM、HEX、ROX和CY5通道荧光信号。
1.3.2.3结果判读 43个间隔序列有对应的荧光与熔点范围,实验运行结束后,分析软件根据每个样本的荧光与熔点组合,自动给出一个对应的43位的二进制码。待测样本,按以下规则判读结果:①优先判读HEX通道的内控熔解峰(IC,44 ℃~48 ℃),如果无内控峰则判定该样品无效,即不含结核分枝杆菌基因组DNA或浓度很低达不到检测要求。②若对应的熔点范围内有熔解峰,则该间隔子存在,表示为“n”;若无熔解峰,则该间隔子缺失,表示为“o”;最后的结果按间隔序列Sp1→Sp43的顺序输出。
1.3.3全基因组测序分析
1.3.3.1全基因组测序 由北京诺禾致源生物有限公司完成检测,利用高通量二代Illumina 测序技术平台,进行DNA 片段文库构建,双端测序,PE150,深度至少200X,每个样品总测序深度数据量≥1G clean data。测序数据经过过滤处理,去除包含Adapter的序列及低质量数据,得到的Clean Data用于后续分析。
1.3.3.2基于全基因组序列对结核分枝杆菌分离株Spoligotying分析 将每株菌株全基因组原始数据通过python2.7软件进行处理,再经过Blastn软件进行序列比对提取出43个间隔序列,每个菌株的43个特定间隔序列结果转化为二进制(0和1)形式输出[4]。
1.4数据分析 将Spoligotying结果数据与SITVITWEB数据库进行比对,获取间隔区寡核苷酸国际型别(spoligotype intemationaltype,SIT)编号,通过PHYLOViZ 2.0软件[5]和https://www.miru-vntrplus.org/MIRU/index.faces网站对结果进行聚类分析并构建进化树,在https://itol.embl.de网站对进化树进行修饰。
2 结 果
2.1WGS和McSpoligotying两种方法检测结果一致性 本研究有361株菌株通过WGS和McSpoligotying两种方法进行结核分枝杆菌Spoligotying基因分型,以WGS方法作为对比方法,McSpoligotyping结果一致性为94.18%(340/361),有21株结果不一致。361株菌株共检测15 523间隔序列,其中21株结果不一致表现在25个间隔序列,不一致率为0.16%(25/15 523),不一致较多间隔序列在第5间隔,有4株菌株出现2个间隔序列不一致,其余标本均为1个间隔序列不一致,具体见表1。

表1 21株MTB间隔序列不一致分布情况Tab.1 Inconsistent distribution of interval sequences in 21 MTB strains
2.2949株MTB McSpoligotying基因分型结果 949株菌株中新发现基因型为121株,已定义基因型为828株。其中已定义基因型共分为84种基因型,主要归属于5种基因家族(BEIJING、T、H、EAI、LAM)和3个家系(Lineage 1、Lineage 2、Lineage 4)[6]。在5种基因家族中BEIJING为60.48%(574/949)、T为21.81%(207/949) 、H为3.69%(35/949)、EAI为0.95%(9/949)、LAM为0.33%(3/949),主要流行基因家族为BEIJING和T。在3个家系中Lineage 1为0.95%(9/949)、Lineage 2为60.48%(574/949)、 Lineage 4为25.82%(245/949)、未定义家系为12.75%(121/949)。具体分型结果见表2和图1。375株非北京基因型和新发现基因型构建进行树及各种基因类型在聚类图中分布情况见图2。

图2 375株非北京基因型和新发现基因型菌株McSpoligotyping分型结果在聚类图中分布情况Fig.2 Distribution of McSpoligotyping genotypes in a cluster map of 375 non-Beijing strains and newly discovered genotype strains
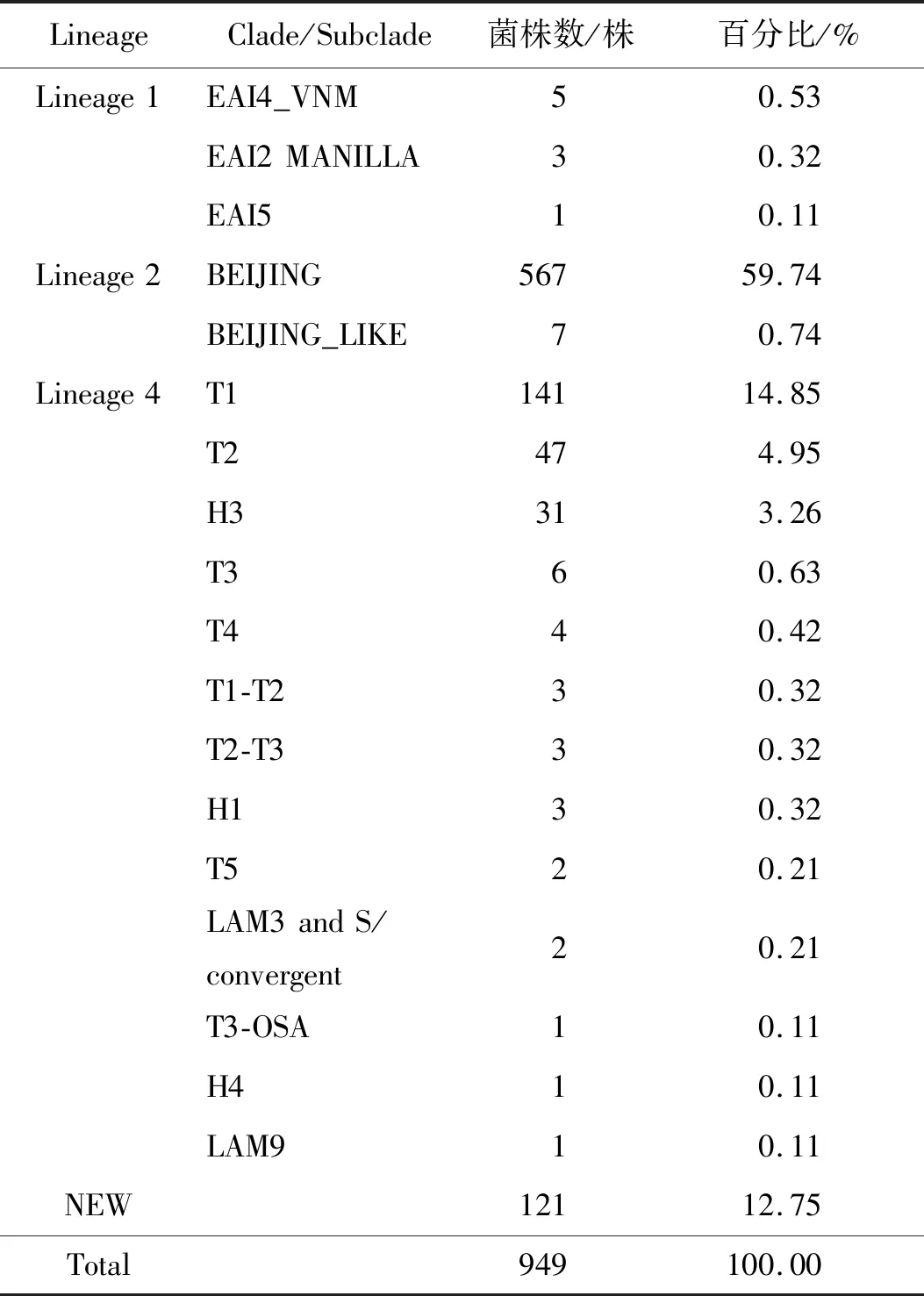
表2 949株MTB McSpoligotyping基因分型结果Tab.2 McSpoligotyping genotyping of 949 MTB strains
2.3成簇性分析 949株菌株中的822株聚为MTB归类于49个簇(2~528株),最大一簇为Beijing家族SIT1基因型,其余的有127株表现为独特的基因型。主要流行簇分别为SIT1(55.64%,528/949)、SIT53(8.49%,80/949)、SIT52(2.21%,21/949)、SIT190(1.90%,18/949)、SIT50(1.69%,16/949)和SIT249(1.58%,15/949),分属于BEIJING、T1、T2和H3 基因家族,各个簇在McSpoligotyping分型结果中占比情况具体见表3。

表3 49个簇菌株在McSpoligotyping分型结果中占比情况Tab.e.3 proportion of 49 cluster strains among the McSpoligotyping genotypes
3 讨 论
间隔区寡核苷酸分型(spoligotyping)是1995年由Kamerbeek J等[7]学者提出来,基于其标准化操作程序和可数字化的结果,多年来spoligotyping是研究结核分枝杆菌遗传多态性和分子流行病学一种有价值的基因分型方法[8]。传统膜杂交spoligotyping方法是利用PCR方法扩增间隔序列,扩增产物与固定在杂交膜上的 43 个合成的间隔序列捕获探针杂交,根据显影结果判读相应的间隔序列是否存在,进而得到菌株的基因型特征。其操作步骤多且非常繁琐,整个检测过程耗时约8 h,结果需要人工判读,容易出现样本污染和判读错误现象。近年来,不少学者提出完善传统spoligotyping的方法。Honisch C等[9]学者利用MALDI-TOF MS平台开发一种基于设计多对引物扩增,无需杂交,耗时约7 h。Ruettger A等[10]学者采用ArrayStrip平台替代尼龙膜杂交,简化了操作步骤和结果记录过程,并能自动化处理信号和数据,耗时约4 h。2013年Ocheretina O[11]利用Luminex Magplex平台在杂交过程采用磁性标记微球体,耗时约4 h。这些所谓的改善方法并未简化PCR扩增后的一系列反应如杂交、显影等,并且要求额外的设备,增加了实验的复杂性和费用。

注:图中的每个圆球及每种颜色均代表一种基因型,圆球内的数字为SIT号;圆球的大小表示相应的基因型所含的菌株数。图1 949株MTB McSpoligotyping基因分型结果在聚类图中分布情况Fig.1 Distribution of McSpoligotyping genotypes in a cluster map of 949 MTB strains
近两年,曾小红等[12]学者提出了一种新型的闭管检测的spoligotyping方法,称之为Mcspoligotyping。Mcspoligotyping是利用多色探针熔解曲线分析技术,仅需一步加样操作即将DNA模板加到PCR反应管中,上机检测仅需两个多小时,分型结果由仪器自动判读。和传统spoligotyping方法相比较省略了PCR扩增后的一系列操作,避免了产物易污染、人工判读错误等缺点。一批次可同时检测30个样本,整个过程耗时约3 h。曾小红等利用来自4个结核病临床实验室收集的1 968份MTB样本,对Mcspoligotyping方法进行多地区大样本量的临床评价,评估该方法的可行性。Mcspoligotyping和传统膜杂交spoligotyping分型结果一致性达99.93%,结果表明Mcspoligotyping方法可在常规实验室使用。本研究对361株菌株进行分型结果一致性为94.18%,有而间隔序列一致性为0.16(25/15 523),不一致较多间隔序列分别在第5间隔,有4株菌株出现两个间隔序列不一致,其余标本均为1个间隔序列不一致。出现不一致主要原因是间隔序列的变化会导致熔解峰转移到一个较低的Tm区域,从而出现假阴性结果和/或假阳性结果。有学者报道[12]认为几乎所有间隔序列不一致结果都是由间隔中的SNPs或插入引起的,由于间隔序列具有高度保守性,间隔序列这种改变也是比较少见。其次,PCR实验室污染也有可能导致假阳性的结果。从本次研究结果可看出,该方法检测结果不影响对结核病疫情处置和分子流行病学研究。
研究发现BEIJING基因家族与云南(55.72%)相当,低于福建(67.23%)、河南(83.53%)、陕西(86.21%)、北京(82.00%)[13-17],表明BEIJING基因家族是我国的主要流行基因型,但地域性分布差异非常大,南方地区所占比例低于北方地区。而国家间的基因型分布差异也很大,东南亚国家[18]主要流行EAI和CAS,欧美地区[19]主要流行LAM和T,邻国缅甸[20]主要流行BEIJING、EAI和LAM。
而非北京基因家族和新发现基因型占39.52%(375/949),这类菌株存在高度的遗传多态性(图2)。非北京基因家族共有80个基因型,其中50个为独立基因型。同时研究发现有121株为新基因型,归属于86个基因型,其中74个为独立基因型,暂未被Spol DB4.0数据库收录。提示广西结核分枝杆菌遗传多态性非常丰富,是研究结核病进化和传播、耐药基因、疫苗等的理想现场。
本研究对成簇性进行分析,发现949株菌株中的822株聚为MTB归类于49个簇(2~528株),成簇率为86.62%(822/949),有127株菌表现为独特的基因型。在成簇的基因型中,BEIJING基因家族占比最大,与罗丹[21]报道的56.95%接近,低于北方的一些省份(82.18%)[22],但仍是广西目前最主要的流行基因型,提示BEIJING基因家族有可能具有更强的传播力,有待进一步研究证实。在49个簇中,大部分簇中每个簇的菌株来源相同一个地方,说明结核病传播大多数可能是近期传播造成。因此,应开展早期快速诊断和治疗,切断传播源,防止结核病疫情进一步传播。
综上所述,本研究利用Mcspoligotyping方法对949株结核分枝杆菌进行基因分型。初步了解到广西地区结核分枝杆菌基因遗传分布和成簇情况,提示广西地区结核分枝杆菌存在高度的遗传多态性和结核病传播情况。Mcspoligotyping方法操作简单、耗时短、费用低,大部分地区均可开展,对结核病疫情以及结核病分子流行病学研究具有重要意义。
(志谢:厦门大学生命科学学院许晔和曾小红博士对本研究给予了科学指导。)
利益冲突:无
猜你喜欢
数学小灵通(1-2年级)(2020年11期)2020-12-28
基层中医药(2020年5期)2020-09-11
小学生学习指导(低年级)(2019年3期)2019-04-22
基层中医药(2018年5期)2018-08-31
特别健康(2018年4期)2018-07-03
中国医学装备(2016年6期)2016-12-01
制造技术与机床(2015年10期)2015-04-09
中国当代医药(2015年36期)2015-03-11
读写算·小学低年级(2014年4期)2014-07-24
西南军医(2014年4期)2014-03-03
