
1例骨髓增生异常-骨髓增殖性肿瘤不能分类型的分析
2021-04-28 01:39税国顺何代莉
国际检验医学杂志 2021年8期
税国顺,黄 莺,何代莉
1.重庆市荣昌区妇幼保健院检验科,重庆 402460;2.重庆永荣矿业有限公司总医院检验科,重庆 402460
骨髓增生异常-骨髓增殖性肿瘤(MDS-MPN)是一组临床表现、实验室检查和细胞形态特征上既有骨髓增生异常综合征(MDS)表现又有骨髓增殖性肿瘤(MPN)表现的髓系肿瘤[1]。在骨髓形态学诊断实际工作中往往接触的是不同的患者,因遗传学和生物学差异,其预后、治疗方案、分子易感性及转归可能完全不同。按世界卫生组织(WHO)分类,综合临床表现、细胞形态学、染色体、流式细胞、基因指标等[2]信息做出准确的诊断,对临床有较好的指导作用。骨髓增生异常-骨髓增殖性肿瘤不能分类型(MDS-MPN,U)是WHO在1999年提出的一个新病种,目前国内报道不多。经中国知网(CNKI)搜索查询,2013年至今,MDS-MPN其他类型多有报道。黎建云等[3]、葛仁英等[4]、吴学琼等[5]对MDS-MPN,U进行了报道。笔者现将1例MDS-MPN,U患者的诊治过程报道如下。
1 临床资料
女,62岁,农民,头昏、乏力1周,晕厥1 h于2019年11月13日11:52入院。既往史:近半个月来一直牙疼不适,在外口服药物治疗。查体:体温36.8 ℃,脉搏78次/分,呼吸22次/分,血压131/74 mm Hg,发育正常,营养中等,形体正常,急性面容,面色苍白,神志欠清,呼之能应,对答稍差,抬入病房,查体合作。嘴唇无发绀,心、肺、腹部未见阳性体征,头颅无畸形,左前额部见约4 cm×4 cm的血肿,高出皮肤,皮肤无破溃,触压痛明显;双眼无充血、压痛,无上睑下垂,巩膜无黄染,双侧瞳孔等大等圆,直径约0.35 cm,对光反射存在。听力良好,鼻腔通畅,耳鼻无出血及脑脊液漏。四肢肌力正常,肌张力不高。病理反射:双侧霍夫曼征、巴宾斯基征、查多克征、奥本海姆征、戈登征阴性。脑膜刺激征:颈软无强直。布鲁辛斯基征及双侧克尼格氏征阴性。
入院诊断如下。(1)晕厥待查:①短暂性脑缺血发作?②脑梗死?③脑出血?④低血糖晕厥?⑤心源性晕厥?⑥其他?(2)左额部皮下血肿。
诊疗经过如下。入院后完善相关检查,头颅CT显示:(1)双侧基底节区及半卵圆中心多发性腔隙性脑梗死;(2)脑萎缩,深部脑白质脱髓鞘改变;(3)左额部皮下血肿。腹部彩超显示:(1)心脏各腔室大小正常;(2)主动脉瓣局限性反流;(3)左室舒张功能减退。颈部血管彩超示:(1)双侧颈动脉内膜粗糙,左侧颈内动脉走行稍弯曲;(2)双侧椎动脉未见明显异常。经颅多普勒显示:左侧大脑中动脉、大脑前动脉血流速度增快。血液相关指标检查结果见表1。
11月13日第1次血细胞分析手工分类200个有核细胞,中性粒细胞(NEU)58.0%,单核细胞(MON)2.0%,杆状核细胞6.0%,幼稚粒细胞24.0%,晚幼红细胞5.0%,网织红细胞(Ret)5.4%。直接抗人球蛋白试验弱阳性。肝功能正常,肾功能正常,血糖(GLU)9.41 mmol/L,凝血4项筛查正常,D-二聚体(D-Dimer)2.30 mg/L,乳酸脱氢酶(LDH)1 084 U/L、α-羟丁酸脱氢酶(α-HBDH)835 U/L、心肌肌钙蛋白I(cTnI)阴性,降钙素原(PCT)0.30 ng/mL。入院后予头孢米诺抗感染,血塞通改善循环,奥拉西坦营养脑细胞。请血液科主任会诊考虑白血病可能性,于2019年11月14日转血液科进一步治疗。予A型RH(D)阳性红细胞悬液共4 U纠正贫血。行骨髓形态学检查:骨髓增生明显活跃,粒细胞系统(以下简称粒系)占79.50%,红细胞系统(以下简称红系)占11.75%,粒红比例为6.77。粒系异常增生,原始细胞比例增高,占11%,后期细胞各阶段可见,形态可见S颗粒、发育差、假性P-H及环形核等核畸形变现象,嗜碱性粒细胞比例增高。红系增生活跃,以中晚幼红细胞增生为主,形态可见芽孢、花瓣等核畸形变现象。环片一周见巨核细胞650个,产板巨核细胞254个,血小板少见。小巨核及多圆核巨核细胞易见。铁染色:细胞外铁++,细胞内铁粒幼细胞占比21%,未见环形铁粒幼细胞。血常规:白细胞计数增高,粒系前体细胞易见,嗜碱性粒细胞比例增高,血小板少见,无核红细胞形态大小不一。考虑急性髓系增殖性病变,建议到上级医院做MIGM分型检查。
陆军军医大学第二附属医院全军血液病中心骨髓细胞形态学检查报告显示,粒巨两系异常增生,伴三系病态造血,提示髓系肿瘤,类型倾向于MDS-MPN,建议做相关BCR/ABL融合基因及染色体核型分析。骨髓病理检查报告显示,骨髓增生极度活跃,幼稚单核细胞较易见,请结合MIGM分型鉴别MDS-MPN或急性髓系白血病(AML)。AML、MDS、MPN免疫学分型流式报告示:淋巴细胞(LYM)占有核细胞(NC)的5.53%,为成熟TBNK细胞;NEU占NC的79.45%,比例正常,CD13/CD16可见发育异常,CD15减弱CD56+。MON占NC的0.42%,比例减低。红系及碎片红细胞占NC的8.73%,比例正常。血型糖蛋白A+可见CD71+。嗜酸性粒细胞占NC的0.17%,嗜碱性粒细胞占NC的0.09%,CD38+DR-CD11c+CD123++CD22+。前体细胞群B占NC的3.82%,CD34+CD117+DR+CD7+CD13+CD38++CD123+CD33+CD11c+MPO-为异常髓系体细胞。意见:异常髓系前体细胞增生伴粒系发育异常,考虑MDS或MDS-MPN。遗传学荧光原位杂交(FISH)分析报告显示,BCR/ABL融合基因阳性细胞占比0.0%。分子生物学血液病相关基因PCR检测报告显示,WT1基因数为1 050,ABL基因数为560 000,WT1/ABL为0.19%,BCR-ABL基因数为0,ABL基因数为710 000、BCR-ABL/ABL基因数为0(BCR-ABL基因是BCR-ABLP210和BCR-ABLP190混合型)。血液病相关基因TEL-ABL、TEL-JAK2、AML1-MDS1/EV11/MTG16、MLL-AF4、MLL-AF9、MLL-AF6/AF10/ELL/ENL、MLL-AF17/AF1q/AF1q/A FX/SEPT6、F1P1L1-PDGFRA、ETV6-PDGFRA、TEL-PDGFRB、NUP98-HoxA13/HoxC11/HoxD13/HoxA9HoxA11/PMX1、BCR-ABL、AML1-ETO、C BFβ-MYH11、PML-RARa、PLZF/STAT5b-RARa、F1P1L1/PRKR1A/NUMA/NPM-RARa、DEK-CAN、N PM-MLF1、E2A-PBX1、E2A-HLF、SIL-TAL1、TEL-AML1全部阴性。按照WHO提出的形态学、免疫学、遗传学、分子生物学结合的MIGM诊断模式,本病例诊断为MDS-MPN,U。患者的骨髓象见图1。
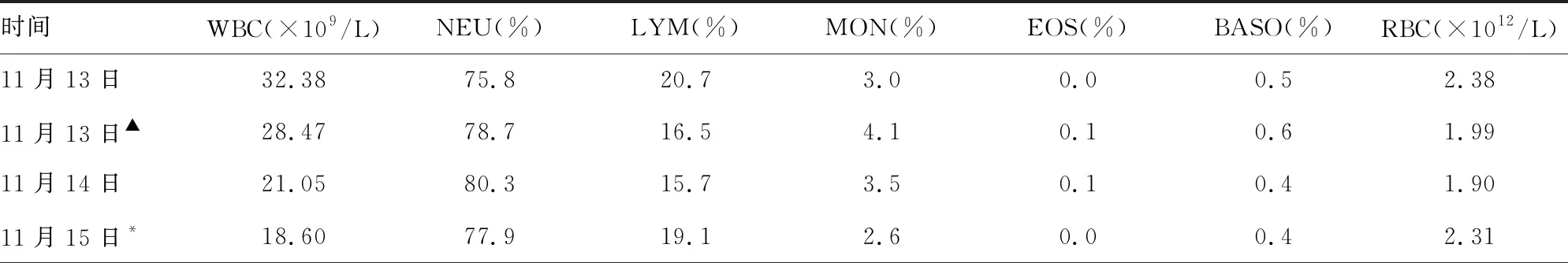
表1 患者不同时间的血液相关指标的检查结果
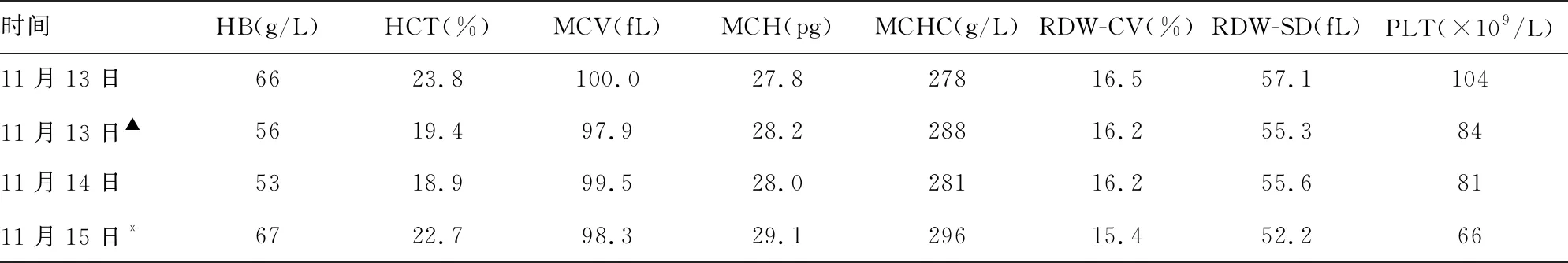
时间HB(g/L)HCT(%)MCV(fL)MCH(pg)MCHC(g/L)RDW-CV(%)RDW-SD(fL)PLT(×109/L)11月13日6623.8100.027.827816.557.110411月13日▲5619.497.928.228816.255.38411月14日5318.999.528.028116.255.68111月15日*6722.798.329.129615.452.266
2 治 疗
入院后予头孢米诺抗感染,血塞通改善循环,奥拉西坦营养脑细胞。输注红细胞悬液纠正贫血。明确诊断后,患者及家属放弃进一步治疗。随访后该患者5个月后病逝。
3 讨 论
MDS是起源于造血干细胞的一组异质性髓系恶性克隆性疾病[6],主要特征是髓系细胞发育异常,表现为无效造血、难治性血细胞减少,具有高危向AML转化的风险[7]。WHO在2008年提出的分型方案分为难治性血细胞减少伴单系发育异常(RCUD),包括难治性贫血(RA)、难治性中性粒细胞减少(RN)、难治性血小板减少(RT)、难治性贫血伴环状铁粒幼红细胞(RARS),难治性血细胞减少伴多系发育异常(RCMD),难治性贫血伴原始细胞增多-1(RAEB-1),难治性贫血伴原始细胞增多-2(RAEB-2),MDS-末分类(MDS-U),MDS伴单纯5q-。
MPN是一组起源于造血干细胞,一系或多系髓系细胞(包括红系、粒系和巨核系)增殖为主要特征的克隆性造血干细胞肿瘤,骨髓有核细胞增多,增殖的细胞可向终末分化成熟,多不伴发育异常;外周血一系或多系细胞增多,外周器官浸润,常伴有肝脾肿大。MPN包括慢性髓细胞白血病(CML)、真性红细胞增多症(PV)、原发性骨髓纤维化(PMF)、原发性血小板增多症(ET)、慢性嗜酸性粒细胞白血病(CEL)、慢性中性粒细胞白血病(CNL)和肥大细胞增多症[8]。
MDS-MPN患者中具有高白细胞数、贫血或血小板减少(也可增加),以及不同程度病态造血特征。骨髓和外周血原始细胞百分数通常<20%。尽管常见脾肿大,但患者临床常表现为MDS或MPN[9]。WHO在2016年提出MDS-MPN分类如下:慢性粒单细胞白血病(CMML);不典型CMML,BCR-ABL1阴性(aCML);幼年型粒单核细胞白血病(JMML);MDS-MPN伴环形铁粒幼细胞和血小板增多;MDS-MPN,U[10]。近年来,随着二代测序在临床的应用,MDS-MPN的基因突变谱系得以解析,从而诊断较前更为精准。
MDS-MPN,U是WHO在1999年提出的一个新病种,由于历史尚短,在许多方面还需要认识和充分的理解。MDS-MPN,U为初诊时,临床、实验室和形态学上既有MDS也有MPN特征,符合MDS-MPN诊断标准,但又不符合CMML、JMML或aCML诊断条件者。特征为一系或一系以上血细胞增多,表现为血小板增多(≥450×109/L),或白细胞计数增加(≥13×109/L),伴有或不伴有明显的脾脏肿大,和未伴有MDS或MPN病史。在MDS特征中,骨髓一系或二系细胞异常增生,表现在外周血中一系或二系血细胞减低;在MPN特征中有一系或多系病态造血,在外周血中为一系或多系血细胞增高。近期没有使用细胞毒药物或生长因子治疗可以解释相关MDS-MPN特征的病史。无BCR-ABL1,也无PDGFRA、PDGFRB或FGFR1基因重排,无t(3;3)(q21;q25)或inv(3)(q21q25)。
黎建云等[3]报道的2例患者中白细胞均升高(33.6×109/L、16.8×109/L),脾脏都有进行性增大。1例血小板正常(112×109/L),年龄23岁,既往有5年苯接触史。1例血小板降低(59×109/L)、年龄43岁。葛仁英等[4]报道的2例患者中,1例白细胞升高(24.1×109/L),血小板降低(90×109/L),脾肿大,年龄47岁;1例白细胞降低(1.58×109/L),血小板升高(633×109/L),肝脾肿大,年龄70岁。吴学琼等[5]报道的1例患者白细胞正常(9.5×109/L),血小板异常升高(1 359×109/L),初诊肝脾未见肿大,年龄69岁。这3篇研究报道的病例其临床、实验室和形态学上既有MDS的特征也有MPN特征,符合MDS-MPN诊断标准,但又不符合CMML、JMML或aCML诊断条件。CLARA等[11]指出,生存率低的患者血小板减少发生的可能性较大,可能代表更具侵袭性的表型。
本例患者发病近期无细胞毒性药物或生长因子治疗病史,肝脾无肿大。血细胞分析结果显示:白细胞计数增高,呈增殖性;血红蛋白、红细胞、血小板未输血前呈进行性下降,重度贫血,红细胞平均体积偏大,血小板明显减少。形态学上粒细胞病态造血特征明显,以粒系病态造血为主,红系和巨核系也有病态表现,符合MDS-MPN的特点;未见环形铁粒幼细胞(可与MDS-MPN伴环形铁粒幼细胞和血小板增多鉴别);免疫学方面,流式细胞学检测可见粒系异常发育,也可以见到异常表型的异常原始细胞,MON占NC的比例为0.42%,比例减低(可与CMML、aCML、JMML鉴别)。遗传学方面,BCR/ABL基因阴性;分子生物学相关基因检测全部阴性。卢兴国等[12]指出,一些之前没有发现MPN慢性期,初诊时已经转化为MDS的患者,如果不能确定MPN病史,归类为MDS-MPN,U是适当的。所有这些都支持本例患者MDS-MPN,U的诊断。
MDS-MPN,U是特异性最差的MDS-MPN亚型,发病率尚不清楚,但约占所有髓系恶性肿瘤的5%,年龄中位数为71岁,其他共同特征包括脾肿大,单核细胞计数低,20%~30%的患者JAK2V617F阳性,且目前还没有公认的MDS-MPN,U的特定细胞遗传学或分子特征,而MDS-MPN,U的细胞遗传学研究主要用于排除其他类似的疾病[11]。
MDS-MPN,U患者少见,迄今尚无共识治疗方案。可依据患者个体情况选用去甲基化药物、免疫调节剂 (如来那度胺)、羟基脲等降细胞药物治疗,在临床实践过程中注意个体化评估和处理。如有合适供体且患者自身状况允许可以考虑造血干细胞移植(HSCT)[13]。随着发病分子机制的不断阐释,分子靶向治疗有望改善这类患者的整体疗效。
猜你喜欢
中老年保健(2021年5期)2021-08-24
中老年保健(2021年6期)2021-08-24
中老年保健(2021年11期)2021-08-22
中华养生保健(2020年3期)2020-11-16
国际放射医学核医学杂志(2020年3期)2020-07-27
医学新知(2019年4期)2020-01-02
文苑(2018年18期)2018-11-08
现代检验医学杂志(2016年2期)2016-11-14
现代检验医学杂志(2016年5期)2016-08-20
中华老年多器官疾病杂志(2016年9期)2016-04-28
