
改进的(S)-3,3'-二(吡咯烷甲基)-八氢联萘酚合成方法
2021-04-28 02:43符世玮汤佳凡古双喜
武汉工程大学学报 2021年2期
符世玮,葛 锐,汤佳凡,明 巍,古双喜
武汉工程大学化工与制药学院,湖北 武汉 430205
1,1'-联萘酚(1,1'-Bi-2-naphthol,BINOL,图1)及其衍生物是一类重要的手性分子,已经广泛应用于不对称合成,分子识别和手性材料中[1-6]。与 BINOL(1,图1)相比,八氢联萘酚(H8BINOL,图1)增强了空间体积并提高了联芳基单元上的电子密度,这使得H8BINOL 衍生物在不对称催化中有着重要的应用[7]。一些3,3'-二取代的H8BINOL,例如3,3'-二(吡咯烷基甲基)-H8BINOL((S)-3,图1),作为高对映选择性催化剂已用于乙烯基[8]或芳基[9]与醛的不对称加成反应,可高收率制备具有高光学纯度的手性醇[10-11],而且,(S)-3 也已用于手性荧光探针的制备[12]。
2010 年报道的合成 3,3'-二取代-H8BINOL 的方法通常是以H8BINOL 为原料的多步类曼尼希反应[9,13]。此类方法存在原子经济性差,操作程序繁琐,后处理过程中污染严重以及溶剂难以回收处理等问题(图2)。这种方法是首先将吡咯烷和多聚甲醛的混合物在0 ℃下搅拌1 h,接下来在室温下搅拌1 h,然后加热到70 ℃,反应过夜,生成吡咯烷基甲醇。最后,在氮气保护下将过量的吡咯烷基甲醇[摩尔数为(S)-2 的 58 倍]与(S)-H8BINOL 在脱气的二噁烷溶液中于60 ℃下反应15 h,以 70%的收率得到(S)-3。2015 年,Gu 等[14]开发了一种简便高效从H8BINOL 一锅合成(S)-3的方法(图3),克服了上述大多数问题。在该方法中,通过柱层析法或重结晶纯化(S)-3,尽管收率较高,但是反应溶剂氯仿具有剧毒,且作为管制品不易购买。本文中,我们报道了从廉价易得的(S)-BINOL 作为起始原料通过两步反应在绿色溶剂乙醇中合成(S)-3 的优化方法[9,13]。

图1 (S)-BINOL 和(S)-H8BINOL 及其衍生物的结构Fig.1 Structures of(S)-BINOL,(S)-H8BINOL and(S)-3

图2 2010 年报道的(S)-3 合成方法Fig.2 Synthetic method of(S)-3 reported in 2010

图3 2015 年报道的(S)-3 合成方法Fig.3 Synthetic method of(S)-3 reported in 2015
1 实验部分
1.1 试剂与仪器
(S)-BINOL、多聚甲醛、四氢吡咯和质量分数10%Pd/C催化剂均购自上海阿拉丁生化技术有限公司。核磁共振光谱(NMR)光谱使用600 MHz Bruker核磁共振波谱仪测定,TMS 为内标。通过薄层色谱法(TLC)在254 nm UV 灯下的硅胶板上监测所有反应。在硅胶(粒径38~48 μm)上进行柱色谱分离。比旋光度在Jasco P1020 数字旋光仪上测定。
1.2 实验方法
(S)-2 的 合 成 :首 先 将 10 g(S)-BINOL(34.92 mmol),1.5 g 负载质量分数 10%Pd 的 Pd/C催化剂(Pd 净含量:质量为(S)-BINOL 的1.5%)和60 mL 无水乙醇放入100 mL 的高压釜中,充氮气置换3 次,再充入氢气至釜内压力为5.5 MPa,然后将混合物升温至70 ℃并搅拌13 h。TLC 监测显示反应完成后,将混合物冷却至室温。接下来,过滤催化剂进行回收,并将滤饼用乙酸乙酯(3×20 mL)洗涤。浓缩合并的滤液,得到粗产物,将其通过快速柱层析法(40 g 硅胶粉)纯化,用乙酸乙酯洗脱,直到通过TLC 未检测到产物。浓缩洗脱液,得到白色固体(S)-2,收率为 99.8%(10.26 g)。1H NMR(600 MHz,CDCl3)δ7.05(d,J=8.4 Hz,2H),6.82(d,J= 8.4 Hz,2H),4.63(s,2H),2.75(t,J= 6.4 Hz,4H),2.15-2.31(m,4H),1.67~1.75(m,8H)。13C NMR(150 MHz,CDCl3)δ151.49,137.25,131.09,130.18,118.98,113.07,29.34,27.22,23.14,23.08。
(S)-3 的合成:在氮气保护下将(S)-2(1.70 mmol,0.5 g),多聚甲醛(4.42 mmol,140 mg),吡咯烷(4.42 mmol,0.373 mL)的混合物在无水乙醇(4 mL)中加热至回流。反应完成后(通过TLC 监控),将混合物冷却至室温,并按如下所述方法Ⅰ,Ⅱ和Ⅲ进行后处理。方法Ⅰ:将反应混合物倒入10 ℃的饱和碳酸氢钠溶液(20 mL)中。将沉淀物过滤,并将滤饼用5 ℃的冷水(3×5 mL)洗涤,最后在50 ℃的真空下干燥4 h 以得到产物,收率为97.9%。方法Ⅱ:如文献[14]所述,通过重结晶纯化产物。方法Ⅲ:如文献[14]所述,将产物通过柱色谱法纯化。mp 159~160 ℃(lit.[14]mp 159~160 ℃),(c= 1.00,THF)]。1H NMR(600 MHz,CDCl3)δ11.21(bs,2H),6.71(s,2H),4.02(d,J=13.8Hz,2H),3.64(d,J=13.8 Hz,2H),2.73-2.71(m,4H),2.61-2.63(m,8H ),2.36-2.38(m,2H),2,19-2.21(m,2H),1.66-1.79(m,16H);13C NMR (150 MHz, DMSO-d6)δ152.22,134.43,126.86,125.94,123.66,119.50,57.87,52.73,28.65,26.42,23.22,22.87,22.83。
2 结果与讨论
先前合成(S)-3 的方法(图2)反应条件苛刻,反应步骤复杂,并且需要过量的起始原料,导致原子经济性极低,并且操作费力。另外,在后处理中使用二噁烷会产生大量的含二噁烷的废水,难以处理。2015 年,Gu 和 Pu 等[14]改进了合成路线,通过一步反应制备(S)-3 且有较高收率(图3),此方法将n(吡咯烷)∶n(多聚甲醛)∶[(S)-H8BINOL]从58∶58∶1 降低至 3∶3∶1,通过简便的操作将两步反应简化为一步反应,无需冷却,溶剂脱气和排放大量含二噁烷的废水。然而,这个方法所使用的溶剂是有剧毒且对人体器官有害的氯仿,因此,我们希望找到一种绿色溶剂代替氯仿。考虑到(S)-H8BINOL 是在乙醇中通过加氢反应制得的,我们尝试在乙醇中进行一锅法的类曼尼希反应(图4)。令我们高兴的是,在这种绿色溶剂乙醇中反应进行的很顺利。
如图4 中所示,在乙醇中用Pd/C 作为催化剂将(S)-BINOL 氢化得到(S)-H8BINOL,然后通过一步类曼尼希反应在乙醇中得到(S)-3。对于加氢反应,先前报道的文献采用了几种不同的体系,比如 H2/PtO2/AcOH[15-16],H2/Rh/EtOH[17],H2/Pd/EtOH[18-19],和 H2/Pd/AcOH[20]等。经过对比,我们决定采用Pd/C 作为催化剂在乙醇中进行加氢反应。我们研究了氢化反应的条件,结果如表1 所示。由表1 可知,反应时间在很大程度上取决于Pd 的负载量和搅拌速率。考虑到Pd/C 催化剂的量和反应时间,第5 种反应条件是较为合理的,其需要少量的Pd[相对于(S)-1 为质量分数1.5%]并且13 h 的反应时间是可接受的。此外,催化剂可以循环使用1 次,以确保(S)-1 的完全转化。但是,如果期待反应在8 h 内完成,可选择第4 种反应条件。尽管使用质量百分比为5.0% Pd 的价格更昂贵,但我们发现回收的催化剂可以重复使用5 次。因此,反应条件4 在大规模制备中是优选方法;而在实验室小规模合成中,一般较少回收钯炭催化剂,所以采用条件5(具体操作见实验部分)。

图4 以(S)-BINOL 为原料的(S)-3 绿色合成方法Fig.4 Green synthesis method of(S)-3 from(S)-BINOL
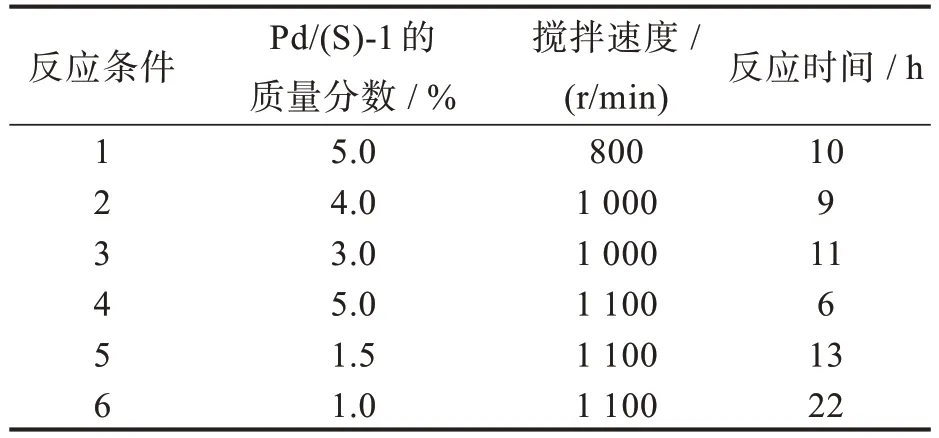
表1 加氢反应条件筛选Tab.1 Screening of hydrogenation conditions a,b
当我们发现类曼尼希反应的一锅法合成可以在乙醇中进行时,我们仍在继续改善反应。如表2所示,我们研究了原料的摩尔比和后处理方法。在这项工作中,我们开发了一种实用的后处理方法(I),该方法不同于先前报道的两种方法Ⅱ(重结晶)和III(柱色谱)。方法Ⅰ比方法Ⅱ和Ⅲ更简洁高效和省时,且收率更高,这是因为饱和碳酸氢钠可降低产物在含乙醇的水中的溶解度。当我们将(S)-2/吡咯烷/多聚甲醛的摩尔比由文献[14]的1∶3∶3 降低为1∶2.6∶2.6 时,收率可高达 97.9%,而继续降低摩尔比至1∶2.4∶2.4 时,收率则有所降低。
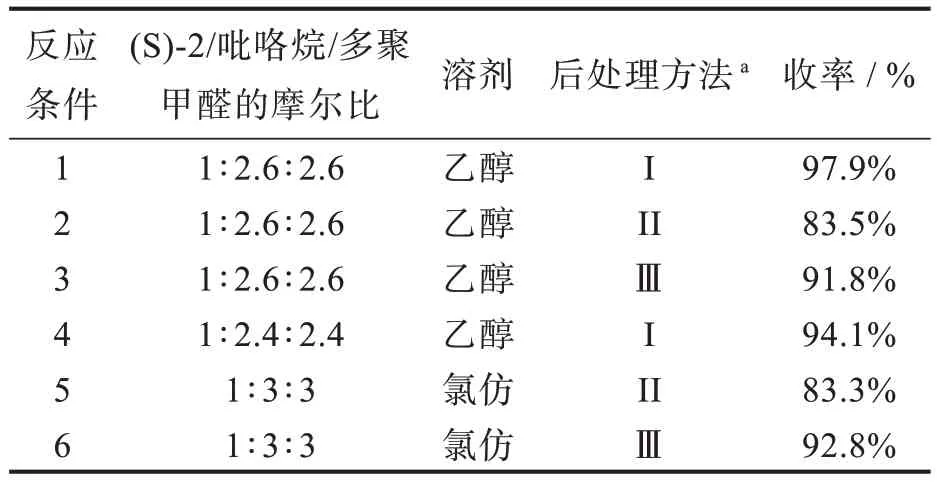
表2 合成(S)-3 的摩尔比和后处理方法的筛选Tab.2 Screening of molar ratios and post-treatment methods for synthesis of(S)-3
3 结 语
总之,我们已经开发了一种实用且环境友好的方法,可通过两个简单的反应从(S)-BINOL 合成(S)-3,产率分别为99.8%和97.9%,这是对本课题组先前开发方法的进一步优化,优势十分明显。在加氢反应中,研究了Pd 的负载量和催化剂的循环利用。在类曼尼希反应中,新方法采用环境友好的乙醇作为溶剂来代替先前报道的氯仿和二噁烷。与以前报道的方法Ⅱ(重结晶)和方法Ⅲ(柱色谱法)相比,制备(S)-3 的新后处理方法更加简洁有效和省时。
猜你喜欢
世界农药(2022年10期)2022-11-10
当代化工研究(2022年11期)2022-06-27
小学阅读指南·低年级版(2022年5期)2022-05-09
当代水产(2021年10期)2022-01-12
能源化工(2021年2期)2021-12-30
今日农业(2020年20期)2020-12-15
能源(2018年10期)2018-12-08
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
能源(2016年10期)2016-02-28
