
高催化活性M-BHT(M=Co,Cu)电催化还原CO2为CH4的密度泛函理论研究
2021-04-17 10:04:46姚会影迟力峰
高等学校化学学报 2021年4期
杨 涛,姚会影,李 青,郝 伟,迟力峰,朱 嘉
(1.苏州大学功能纳米与软物质研究院,江苏省碳基功能材料与器件高技术研究重点实验室,苏州215123;2.北京师范大学化学学院,北京100875;3.南洋理工大学材料科学与工程学院,新加坡639798)
1 Introduction
The electrochemical reduction of carbon dioxide(CO2)into chemicals of economic value(hydrocarbon or alcohols)holds a sustainable and promising way to produce renewable energy sources[1—4]. The best solutions for energy transition from“fossil fuel economy”to a sustainable“CO2economy”are reducing CO2to CH4or alcohol compounds[5]. However,the free electrocatalysts for reducing CO2to these target products are in the exploration and need more development and improvement,particularly theoretical outlooking[6—15]. The most limitation mainly caused by the electronic structure properties of catalysts may affect the product’s selectivity.The apparent evidence is given by the large overpotential needed for CO2reduction to these products[5].Hence,it is critically necessary and essential to explore and predict the catalytic activities and selectivity of the catalysts for CO2reduction reaction(CRR)from the atomic level. So far,various two-dimensional(2D)materials,including metals,metal oxides,chalcogenides,and even metal-free catalysts,have been extensively demonstrated both theoretically and experimentally to have significant potential for CRR[1,13,16—21]. As the most efficient Cu-based material for CRR electrocatalysts,2D Cu-C3N4has excellent catalytic activity[the Gibbs free energy change(ΔGL)of the rate-limiting step for CH4formation is 0.75 eV]for CRR[13]. The critical requirements are the development of highly active electrocatalysts,by increasing the number of the active sites[22]. Recently,metal-BHT(metal=Cu,Co;BHT=benzenehexathiol)[23,24]as a new type ofπ⁃conjugated 2D materials have been synthesized,which has abundant exposed surface atoms and unique electronic properties. For example,Co-BHT is a ferromagnetic half-metal. Moreover,the theoretical studies indicated that Co-BHT shows the spin-filtering effect and strong attractivity to CO[25]. CO is the critical intermediate for CO2reduction to CH4. CO’s adsorption on the catalyst surface indicates it can be further hydrogenated to other reaction intermediates[13,26]. This property suggests that Co-BHT is candidate efficient catalysts for CO2reduction to CH4.
In this work,we select the monolayer Co-BHT and Cu-BHT as electrocatalysts to investigate the reduction mechanism of CO2to CH4from the atomic level and compare their catalytic performance. The reaction mechanisms for CO2reduction into CH4catalyzed by Co-BHT and Cu-BHT were determined by the Gibbs free energy calculations. Thed-band center and ΔGLcalculations suggest that Co-BHT is a more promising candidate for CH4production than Cu-BHT.
2 Computational Method
The spin-polarized electronic structure calculations were carried out by the Viennaab initiosimulation package(VASP)[27,28]. The projector augmented wave(PAW)[29]method was adopted to describe the ionelectron interaction,and the electron exchange-correction was represented by the Perdew-Burke-Ernzerhof(PBE)functional[30]. Regarding the plane-basis set,a kinetic energy cutoff of 500 eV with Gaussian-level smearing of 0.05 eV was used in all calculations. Thek-space samplings were set as 5×5×1 for the Cu-BHT unit cell and 2×2×1 for the Co-BHT model. The convergence thresholds for structural geometry optimization and for electronic structure iteration were set as 0.1 eV/nm and 10−5eV,respectively. According to the previous studies,the Grimme’s method(DFT-D3)[31—35]can obtain more accurate adsorption results for CRR intermediates. So,the DFT-D3 was applied to all calculations to include the van der Waals correction.
The Gibbs free energy(G)of each species was defined as

whereEtotal(eV),EZPE(eV),CP(J·K−1·mol−1),S(J·K−1·mol−1)andT(K)are total electronic energy,zeropoint energy,heat capacity,entropy,and system temperature(298.15 K),respectively. Free energy of the adsorbates was calculated by treating all 3N(Nis the number of atoms of the adsorbate)degrees of freedom of the adsorbates as vibrational without consi-dering contributions from the substrate,in which all vibrations were treated in the harmonic oscillator approximation. TheEZPE,CP,andSwere calculated from these vibrations based on standard methods[36]. The parameters of the molecules were taken from the NIST database[37]. According to previous studies,the adsorbate solvation effects were similar:hydroxyl adsorbates(*OH)and hydroxyl functional groups(*R-OH)were stabilized by 0.50 and 0.25 eV,respectively,and intermediates containing adsorbed CO such as*CO and*CHO were stabilized by 0.10 eV[26]. Gas-phase energetics of species containing a CO backbone were compensated to match experimental values;the total energy corrections for CO2and CO were 0.16 and −0.27 eV,respectively[38]. The Gibbs free energy change of the elementary reaction step(such as*CO+H++e−→*CHO)was calculated as below[26]:
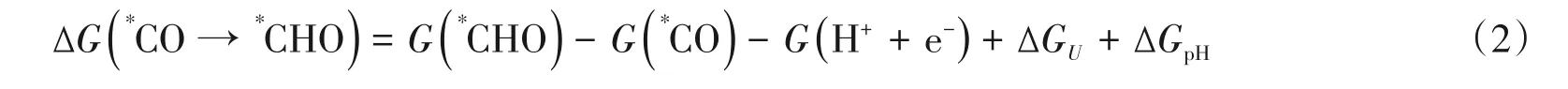
According to the computational hydrogen electrode(CHE)[39]model,theG(H++ e−) is equal to half of the free energy of gaseous hydrogen[1/2G(H2)]at the electrode potential of 0 V(vs. RHE)and all temperature. ΔGU=−neU,wheren(1)is the number of transferred electrons,eis the elementary positive charge,andU(V)is the bias applied on the electrode. ΔGpH(ΔGpH=kBT× ln10 × pH)is the correction of the pH,wherekBis the Boltzmann constant. pH is zero for the acidic condition.
The adsorption energy(Eads)[40]is used to evaluate the binding strength between the adsorbates and the substrate,which is defined as below:

whereEX*(eV),EX(eV)andE*(eV)represent the total energy of the adsorption system,the adsorbate,and the substrate,respectively. According to this definition,the negativeEadsindicates that the adsorption is exothermic process.
Thed-band center(εd)was calculated by the following equation based on projected density of states(PDOS)calculation results as below[41—46]:

wheren(ε)represents the density of states occupied when the electron energy equal toε(eV).
3 Results and Discussion
Cu-BHT and Co-BHT have hexagonal unit cell,as shown in Fig.1. By means of DFT optimization,the 2D lattice parameters of Cu-BHT are obtained to bea=b=0.875 nm,agreeing well with the experimental observations(a=b=0.845 nm)[24]. The 2D lattice parameters of Co-BHT area=b=1.470 nm,which are also in agreement with previous theoretical calculations(a=b=1.471 nm)[25].

Fig.1 Structures of Cu⁃BHT(A)and Co⁃BHT(B)
The total density of states(DOS)and partial density of states(PDOS)of Cu-BHT and Co-BHT are shown in Fig.2. For Cu-BHT,the PDOS shows that the total DOS near Fermi level is contributed by S atoms,C atoms and d-orbital density of states of the Cu atoms. For Co-BHT,the density of states projected onto C,S,and metal atomic orbitals show thatd-orbital density of states of the Co atoms contribute mainly to the total DOS for both spin-up and spin-down channels,while small amounts of the total DOS are from S atoms and C atoms. Thed-band center gives an indication of the catalyst activity,and it is an important indicator for predicting the catalytic activity of metal and half-metal catalysts[43—47]. The closerd-band center to the Fermi level usually leads to better catalytic activity of the catalyst[13,47]. Thed-band centers of Co-BHT for spin-up and spin-down channels are −1.62 and −0.65 eV,respectively,which are closer to the Fermi level than those of Cu-BHT(−2.38,−2.38 eV),indicating that Co-BHT may have stronger interactions with the CO2reduction intermediates.

Scheme 1 Possible reaction paths for CO2 reduction to CH4 by electrochemical catalysis on the catalyst surfaces
According to previous studies[13,26,47~49],there are four possible reaction paths for CO2reduction to CH4,as shown in Scheme 1. Among these four paths,the first step starts with hydrogenation of the physisorption CO2to form*COOH. The second step is*COOH→*CO,which involves the OH desorption and further hydrogenation to form H2O and left*CO on the catalyst surface. The major difference between path Ⅰand paths Ⅱ—Ⅳis that*CO is hydrogenated to*COH or*CHO intermediate. For path Ⅱand path Ⅲ,the major branching point is whether*CHO is reduced to a CHOH or*CH2O intermediate by the formation of an O—H bond or a C—H bond,respectively. For path Ⅲand path Ⅳ,the major branching point depends on whether*OCH3or*CH2OH is formed. Moreover,the major difference between path Ⅲand other paths is that the formation of CH4occurs during the sixth proton/electron pairs transferring process in path III,while lefts adsorption*O intermediate on the catalyst surface,which would be further reduced to*OH. Additionally,the last step of path III is the formation of H2O,while,for the rest paths,the last step is the formation of CH4.
Based on these reaction paths,we first investigated the adsorption performance of all reaction intermediates on Cu-BHT and Co-BHT by density functional theory(DFT)calculations. All possible adsorption sites of these reaction intermediates were considered to determine the most stable sites(Table S1 and Table S2,see the Supporting Information of this paper). The values of the adsorption energy(Eads)of CO2,*COOH,*CO,*CHO and CH4on M-BHT(M=Cu,Co)are summarized in Table 1.

Table 1 Calculated Eads of CO2, *COOH, *CO, *CHO and CH4 on Cu-BHT and Co-BHT
For Cu-BHT,Cu,S and C sites are considered as the adsorption sites[see Fig.1(A)]. The results show that the CO2,*COOH,*CHO,*CH2O,*OCH3,*O,*OH,*CH2OH and CH4prefer to absorb on the S sites,while the*CO and*C prefer to adsorb on the Cu sites,and other reaction intermediates prefer to adsorb on the C sites. In general,the adsorption is physisorption when the absolute value ofEadsis less than 0.31 eV(30 kJ/mol)[50],and the adsorption is strong chemisorption when the absolute value ofEadsis large than 1 eV[51,52].As shown in Table 1,theEadsof CO2and CH4are −0.19 and −0.15 eV,respectively,indicating that the adsorption is physisorption and CO2and CH4can be desorbed from the catalysts. However,theEadsof*CO is−0.41 eV,which indicates that the adsorption is weak chemisorption so that CO can be further hydrogenated.For Co-BHT,the most stable adsorption sites of possible reaction intermediates were obtained by DFT calculations. The calculation results show that the CO2,*COOH,*CHO,*OH,*CH2O,*CH2OH and CH4prefer to adsorb on metal atom sites(Co sites),which are different from the adsorption sites on Cu-BHT(prefer to adsorb on non-metal atom site:S site). TheEadscalculations show that both CO2and CH4are physically adsorbed on Co-BHT,which is the same as that on Cu-BHT(see Table 1). Furthermore,theEadsof*CO is−1.42 eV,indicating that the adsorption of CO on Co-BHT is strong chemisorption,which means that CO can be hydrogenated.
The Gibbs free energy profile of the four reaction paths for CO2reduction to CH4on Cu-BHT is displayed in Fig.3(A). The first and the second proton/electron pairs transferring steps for the four reaction paths are CO2→*COOH and*COOH→*CO with Gibbs free energy changes by 0.76 and −0.17 eV,respectively. The third step is the production of*COH(path Ⅰ)or*CHO(paths Ⅱ—Ⅳ)by hydrogenating*CO. The Gibbs free energy change of*CO→*COH is 0.50 eV,which is larger than that of*CO→*CHO(0.17 eV). Therefore,the reaction of path Ⅰis more difficult thermodynamically than that of the rest reaction paths. The*CHOH intermediate is the key reaction intermediate in path Ⅱ. The ΔGof*CHO→*CHOH is 0.21 eV,while ΔGfor the formation of*CH2O in path Ⅲand Ⅳis 0.04 eV. The result indicates that the*CHO intermediate will be hydrogenated to*CH2O rather than*CHOH. Hence,path Ⅱis not the prior reaction path compared with paths Ⅲand Ⅳ. The major difference between path Ⅲand path Ⅳis the hydrogenation of*CH2O to*OCH3versusto*CH2OH(path Ⅳ). The elementary reaction step of*CH2O→*OCH3in path Ⅲis increased in free energy by 0.17 eV,whereas the ΔGof*CH2O→*CH2OH(in path Ⅳ)is −0.70 eV. Thus,the CO2follows path Ⅳreduction to CH4on Cu-BHT. Additionally,the ΔGof*CH2OH→*CH2reaction is 0.60 eV,which is lower than that of the first elementary reaction step(0.76 eV). Therefore,the path Ⅳpossesses the lowest energy reaction pathway of CO2reduction to CH4on Cu-BHT,and the rate-limiting step is CO2→*COOH with Gibbs free energy change of 0.76 eV[see Fig.3(A)].

Fig.3 Free energy diagrams of CO2 reduction to CH4 on Cu⁃BHT(A)and Co⁃BHT(B)
For Co-BHT,the calculated Gibbs free energy diagram of the possible reaction pathways for CRR is summarized in Fig.3(B). The results show that the steps of CO2→*COOH and*COOH→*CO have ΔGvalue of 0.29 and −0.71 eV,respectively. As shown in Fig.3(B),the ΔGfor the formation of the key intermediate*COH in path Ⅰis 1.15 eV,which is larger than that of the reaction*CO→*CHO(0.66 eV),indicating that path Ⅰis not the lowest energy reaction pathway for CO2reduction to CH4on Co-BHT. Moreover,the ΔGfor*CHO hydrogenation to*CH2O(in paths Ⅲand Ⅳ)is 0.56 eV,which is larger than that of reduction of*CHO to*CHOH(0.01 eV). Therefore,path Ⅱis the lowest energy pathway for CO2reduction to CH4on Co-BHT.
To compare the catalytic mechanism and the performance of Cu-BHT and Co-BHT for CO2reduction to CH4,the calculated free energy diagram of CRR is summerized in Fig.4 and Fig.S1(see the Supporting Information of this paper). The rate-limiting steps of CH4formation,CO2→*COOH with ΔGLof 0.76 eV on Cu-BHT,and*CO→*CHO with ΔGLof 0.66 eV on Co-BHT,indicate that the CO2reduction to CH4catalyzed by Co-BHT is more favorable. Note that ΔGLfor CO2reduction to CH4on Cu(211)[26]and Cu-C3N4[13]are 0.74 and 0.75 eV,respectively. Thus,our calculations show that both Cu-BHT and Co-BHT are promising electrocatalyst candidates for CO2reduction to CH4.

Fig.4 Free energy diagram of the lowest energy pathway of CO2 reduction to CH4 on Cu⁃BHT and Co⁃BHT
4 Conclusions
In summary,we investigated the electrocatalytic activities of Cu-BHT and Co-BHT for the reduction of CO2to CH4based on DFT calculations. The reduction of CO2to CH4on Co-BHT and Cu-BHT follows different reaction pathways and rate-limiting steps. The lowest energy reaction pathway for CO2reduction into CH4catalyzed by Co-BHT is CO2→*COOH→*CO→*CHO→*CHOH→*CH→*CH2→*CH3→CH4,with the rate-limiting step of*CO→*CHO. The reaction pathway on Cu-BHT is CO2→*COOH→*CO→*CHO→*CH2O→*CH2OH→*CH2→*CH3→CH4. The DFT calculations show that the Co-BHT has higher catalytic activity than Cu-BHT for CRR because it possesses small ΔGL(0.66 eV)for CO2reduction to CH4than that of Cu-BHT(0.76 eV). Thed-band center calculations show that Co-BHT may has better catalytic activity than Cu-BHT for CRR,which is consistent with the onset potential calculations.
Acknowledgments
The most calculations were supported by HPC from Key Laboratory of Theoretical and Computational Photochemistry,Ministry of Education,College of Chemistry,Beijing Normal University. Especially acknowledge the discussion and suggestions of M-BHT with Dr. HUANG Xing and Prof. XU Wei.
The Supporting Information of this paper see http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200729.
This paper is supported by the National Natural Science Foundation of China(Nos.21790053,51821002,21773016).
猜你喜欢
无机盐工业(2023年12期)2023-12-19 07:02:42
美育学刊(2023年2期)2023-04-21 12:10:26
寻根(2022年2期)2022-04-17 11:01:38
佛山科学技术学院学报(自然科学版)(2022年1期)2022-02-15 09:35:20
中小学校长(2021年8期)2021-09-11 09:39:54
科学中国人(2019年22期)2019-12-31 05:42:58
校园英语·下旬(2019年8期)2019-10-07 10:06:16
中国机械工程(2018年21期)2018-11-13 08:55:18
校园英语·中旬(2018年6期)2018-09-08 10:21:20
材料科学与工艺(2017年6期)2018-01-08 05:56:06
