
Primary localized gastric amyloidosis: A scoping review of the literature from clinical presentations to prognosis
2021-04-17 06:55XinYuLinDanPanLiXuanSangBingChang
World Journal of Gastroenterology 2021年12期
Xin-Yu Lin, Dan Pan, Li-Xuan Sang, Bing Chang
Abstract
Localized gastric amyloidosis (LGA) is a rare disease characterized by abnormal extracellular deposition of amyloid protein restricted to the stomach and it is confirmed by positive results of Congo red staining. Over decades, only a few cases have been reported and studies or research focusing on it are few. Although LGA has a low incidence, patients may suffer a lot from it and require proper diagnosis and management. However, the pathology of LGA remains unknown and no overall review of LGA from its presentations to its prognosis has been published. Patients with LGA are often asymptomatic or manifest atypical symptoms, making it difficult to differentiate from other gastrointestinal diseases.Here, we report the case of a 70 -year-old woman with LGA and provide an overview of case reports of LGA available to us. Based on that, we conclude current concepts of clinical manifestations, diagnosis, treatment, and prognosis of LGA, aiming at providing a detailed diagnostic procedure for clinicians and promoting the guidelines of LGA. In addition, a few advanced technologies applied in amyloidosis are also discussed in this review, aiming at providing clinicians with a reference of diagnostic process. With this review, we hope to raise awareness of LGA among the public and clinicians.
Key Words: Gastroscopy; Changes of gastric mucosa; Primary localized gastric amyloidosis; Clinical presentations; Prognosis
INTRODUCTION
Amyloidosis is a group of conformational diseases characterized by the accumulative extracellular deposition of insoluble fibrils in various tissues and organs as a result of protein folding disorders[1]. Large deposits may lead to the loss of the normal structure of tissues, subsequent organ dysfunction, and even death. At present, 36 different amyloid fibrils have been identified that are associated with amyloidosis[1]. Amyloid fibrils determine the properties of the amyloid diseases, and the subtypes of amyloidosis are also named after the corresponding fibrils. For example, light chain(AL) indicates that amyloid fibrils are derived from immunoglobulin light chains, and the resulting disease is referred to as AL amyloidosis[2]. According to the amyloid distribution, amyloidosis is divided into systemic and localized amyloidosis. Systemic amyloidosis is universal, while localized amyloidosis is a rare condition that only comprises 12 % of newly identified amyloidosis cases[3].
Amyloidosis is a rare disease, among which AL amyloidosis is the most common type. An epidemiological study in Sweden reported an incidence of nonhereditary amyloidosis of 8 .29 per million person-years, among which AL amyloidosis accounted for 3 .2 per million person-years[4]. A nationwide study in the United States reported an increasing incidence of AL amyloidosis, from 9 .7 per million person-years in 2007 to 14 .0 per million person-years in 2015 [5]. Although rare, amyloidosis can result in a severe disease burden, as reflected by patients’ poor scores on assessments of the health status compared to the general population in a recent study of 341 patients[6].Moreover, without proper intervention, it may ultimately develop into a fatal disease.From 2000 to 2008 , 0 .58 per thousand deaths were due to amyloidosis in England, and its proportion of deaths has doubled, indicating a tendency to increase[7].
Gastrointestinal involvement manifests as systemic amyloidosis (79 %), while it is relatively rare in localized cases (21 %), according to a retrospective study of 76 patients of biopsy-proven gastrointestinal amyloidosis evaluated in 1998 -2011 [8]. Localized gastric amyloidosis (LGA) is an extremely unusual condition. Generally, it refers to amyloidosis confined to the stomach without evidence of potential plasma cell dyscrasia or the involvement of other organs, particularly the heart, liver, kidney, or nerve[3,8]. More specifically, the precursor protein of amyloid is produced and deposited in the stomach without detection in a remote site[9]. According to a retrospective study of gastrointestinal biopsies from 542 patients, the most common amyloid subtype in the stomach is AL (λ), followed by transthyretin (ATTR), AL (κ),and serum amyloid A (AA)[10].
Given the rare reports and unsolved problems associated with LGA, we present a case of LGA (Figures 1 and 2 ) and collect several recent case reports of LGA available in the literature to present its clinical manifestations, diagnosis and differential diagnosis, and treatment. In addition, we will describe our understanding of its pathogenesis.
PATHOGENESIS
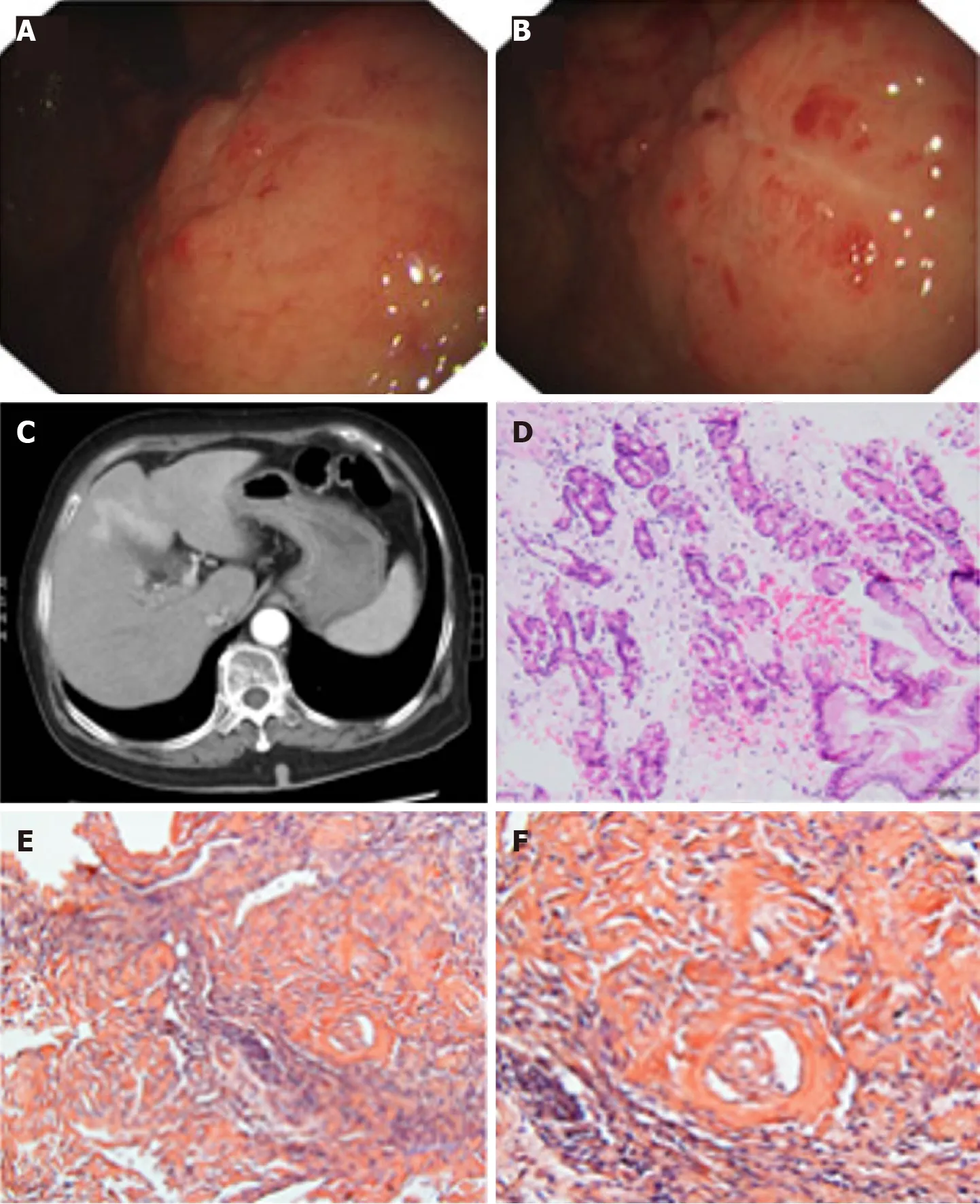
Figure 1 Pathological findings from a 70 -year-old woman with localized gastric amyloidosis. The patient came to the hospital with a chief complaint of hematemesis for 2 wk. A and B: Endoscopic findings show multiple congested fragile ulcers scattered in the gastric body and fundus. A 4 .0 cm × 4 .0 cm area of the mucosa with edema, ulcers, and poorly delineated boundaries was observed in the anterior wall of the gastric body and fundus. The lesion appeared as a rough, congested area with edema, localized superficial fragile ulcers and active bleeding. Spot-like congested erosions exhibited a scattered distribution in the mucus of the sinus; C: CT reflected diffusely thickened gastric walls and shallow folds of the mucosa, while no abnormalities were observed in the enhanced images;D: H&E staining revealed massive amyloid fibrous connective tissues deposited in the interstitium with inflammatory cell infiltration; E and F: Congo red staining confirmed the existence of the amyloid protein (E: Congo red, × 200 magnification; F: Congo red, × 400 magnification).
To date, the pathogenesis of amyloidosis is unclear, but current studies and hypotheses provide a few insights. All amyloid fibrils share the same antiparallel cross-β secondary structure, a structure with a high propensity for self-aggregation, as observed under an electron microscope[11 ,12]. Tightly bound β-sheets form the protofilament, and several protofilaments twist and eventually form amyloid fibrils[13].Proteins may have a potential intrinsic propensity of misfolding that is influenced by multiple factors, such as aging and stably high concentrations in serum[11]. For example, wild-type TTR forms amyloid fibrils in older individuals, even at normal concentrations, while serum amyloid A and β2M only become amyloidogenic at a persistently high concentration[14]. Different amyloid fibrils may be susceptible to different conditions that trigger misfolding. Mutations may induce the formation of amyloidogenic proteins[15 ,16]. Mutations in genes that encode amyloid fibrils, such as TTR, trigger familial amyloidosis[17]. Somatic mutations have been identified in AL amyloid fibrils. The N-terminal strand of the light chain variable domain prevents protein aggregation, and its mutation destabilizes the protein and accelerates light chain fibrillogenesis[18 ,19]. However, the exact relationship between mutations and amyloidosis remains unknown. In addition, further thermodynamic investigations reveal that many environmental factors, such as temperature and pH, are related to the conformational stability of amyloid fibrils and may be critical factors contributing to the conversion of a normal protein into amyloid fibrils[13 ,20 ] (Figure 3 ).

Figure 2 Results of immunochemical staining using several antibodies excluded the diagnosis of gastric cancer. A: CKPAN-positive staining in the glands; B: Periodic Acid-Schiff staining is negative; C: Smooth muscle actin-positive staining in the muscularis mucosa; D: Vimentin-positive staining.
In addition, several protein cofactors, nonfibrillar components, glycosaminoglycans(GAGs), and serum amyloid P (SAP) are present in all amyloid deposits and are believed to function in amyloidogenesis and persistence. GAGs, a main component of the extracellular matrix, are associated with amyloid fibrils in AL amyloidosis, and their size and charge may play an important role in the acceleration and stability of amyloid formation[19]. SAP is a type of plasma glycoprotein that binds to all types of amyloid fibrils in its calcium-bound state and protects them from proteolysis[21]Amyloid fibrils are digested in vitro by proteases and phagocytic cells, while amyloid fibrils likely exhibit relative stability in vivo with the assistance of SAP[22]. According to an in vitro study, SAP may accelerate and stabilize the formation of Aβ42 , the amyloid fibrils responsible for Alzheimer’s disease[23]. The severity of amyloid deposition in SAP knockout mice is decreased considerably[24]. Therapies targeting SAP have been tested and confirmed to exert stable effects[25].
The mechanism by which amyloid deposits damage the organs and lead to dysfunction is unclear. The site of deposition may be related to multiple factors, such as the pH, protein concentration, proteolytic processing, and fibril seeds. Different amyloid fibrils exhibit a preference for specific organs; for example, β2-microglobulin prefers joints[11]. In patients with LGA, amyloid is universally present in the walls of small vessels, and most of these amyloid deposits are classified as AL amyloidosis(12 /22 ). In cardiac amyloidosis, amyloid deposits in small vessels lead to symptoms of cardiac ischemia[26]. In gastric amyloidosis, we are able to detect the same distribution patterns, but no connections have been reported to date. Furthermore, the toxicity of amyloidogenic light chain proteins (AL-LC) may be responsible for this condition. An investigation of the potential mechanism revealed that the injection of human AL-LC within a zebrafish model causes cell death. Human AL-LC induces intracellular oxidative stress and alters the cellular redox status, eventually leading to cardiac dysfunction, which is not attributed to the deposition of amyloid protein[27]. As shown in another study, AL-LC mediates cardiomyocyte apoptosis and dysfunction through the activation of p38 mitogen-activated protein kinases[28]. Lysosomal dysfunction has also been reported to provoke the proteotoxicity of AL-LC by contributing to impaired autophagy[29]. Investigations of the precise molecular mechanism of cardiac amyloidosis are ongoing and may explain how the amyloid protein contributes to organ damage. These studies may reveal the mechanism and inspire further studies of gastric amyloidosis.
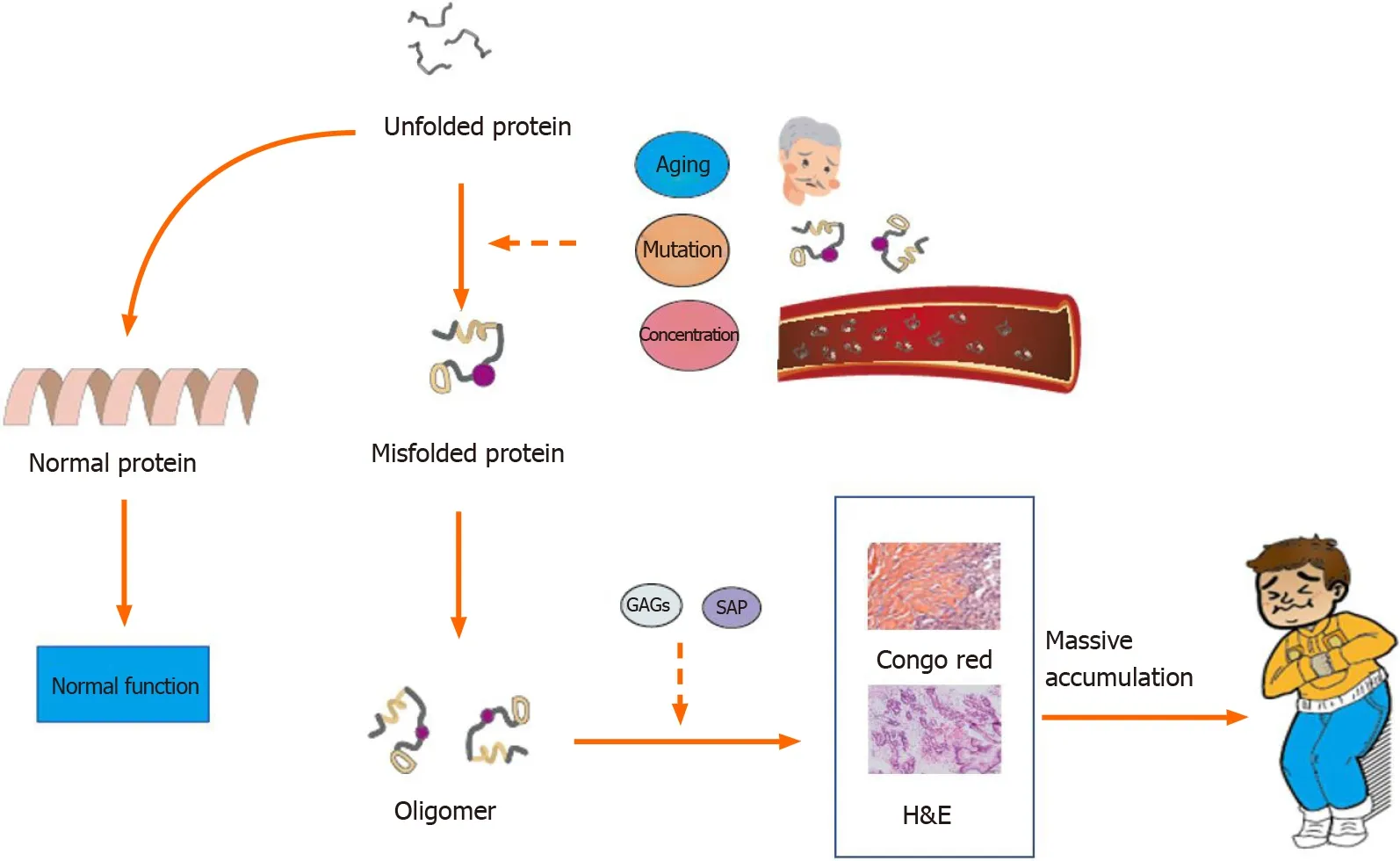
Figure 3 Potential molecular events leading to amyloidosis. Without intervention, the unfolded protein becomes the normal protein. Factors such as aging, mutation, and high blood concentrations may cause protein misfolding. The misfolded protein aggregates into oligomers and forms fibrils with the assistance of glycosaminoglycans and serum amyloid P. Massive deposition of amyloid fibrils leads to amyloidosis. GAGs: Glycosaminoglycans; SAP: Serum amyloid P.
CLINICAL MANIFESTATIONS
LGA mainly targets middle-aged and elderly people aged from 50 to 80 years. Equal numbers of male and female patients are affected, and no sex differences have been detected. The clinical manifestations of LGA often mimic other common gastric diseases and lack specificity, ranging from an asymptomatic disease to epigastric discomfort, pain, weight loss, anemia, heartburn, nausea, hematemesis, tarry stool,fatigue, and other symptoms (Figure 4 ). Generally, these manifestations depend on the sites and extent of amyloid involvement[30]. Patients in our reviewed case studies often presented without a chief complaint, and LGA was generally diagnosed based on the results of tests for other gastric diseases. Among the 22 cases that we reviewed, 13 cases of AL LGA and 1 case of AA LGA were identified. Most AL LGA cases manifest asymptomatically[31 -35 ] or with epigastric discomfort[36 ,37], while AA LGA exclusively manifests as epigastric discomfort[38 ] (Table 1 ). To date, no association between symptoms and amyloid types has been confirmed[39]. Further discussion of whether a correlation exists between clinical presentations and amyloid fibrils in patients with LGA is worthwhile.
DIAGNOSIS
Imaging findings
Endoscopic results have identified variable features in patients. Amyloid deposits mainly invade the gastric body and antrum (Figure 5 ). Generally, the deposits manifest as single or multiple lesions in the form of a mass[40 -43 ], ulcer[38 ,44 ], fold[36 ,45],elevation[37 ,46 ], or submucosal tumor-like feature[34 ,47]. The borders may be clear or unclear. These findings are consistent with those of previous studies. A remote study of 37 patients with gastrointestinal amyloidosis revealed that erosions were the most common presentation in the stomach, followed by granules, ulcers, and mucosal friability[48]. However, a recent study provided a different order of a normal appearance, followed by erythema, erosions, and nodularity[39]. The samples included both localized and systemic amyloidosis, and thus, the difficulty in distinguishing localized amyloidosis from systemic involvement simply based on endoscopic featuresis noted given the similarities in appearance. Furthermore, some patients appear normal in endoscopic evaluations, and endoscopy should therefore not be used for an independent diagnosis[39].
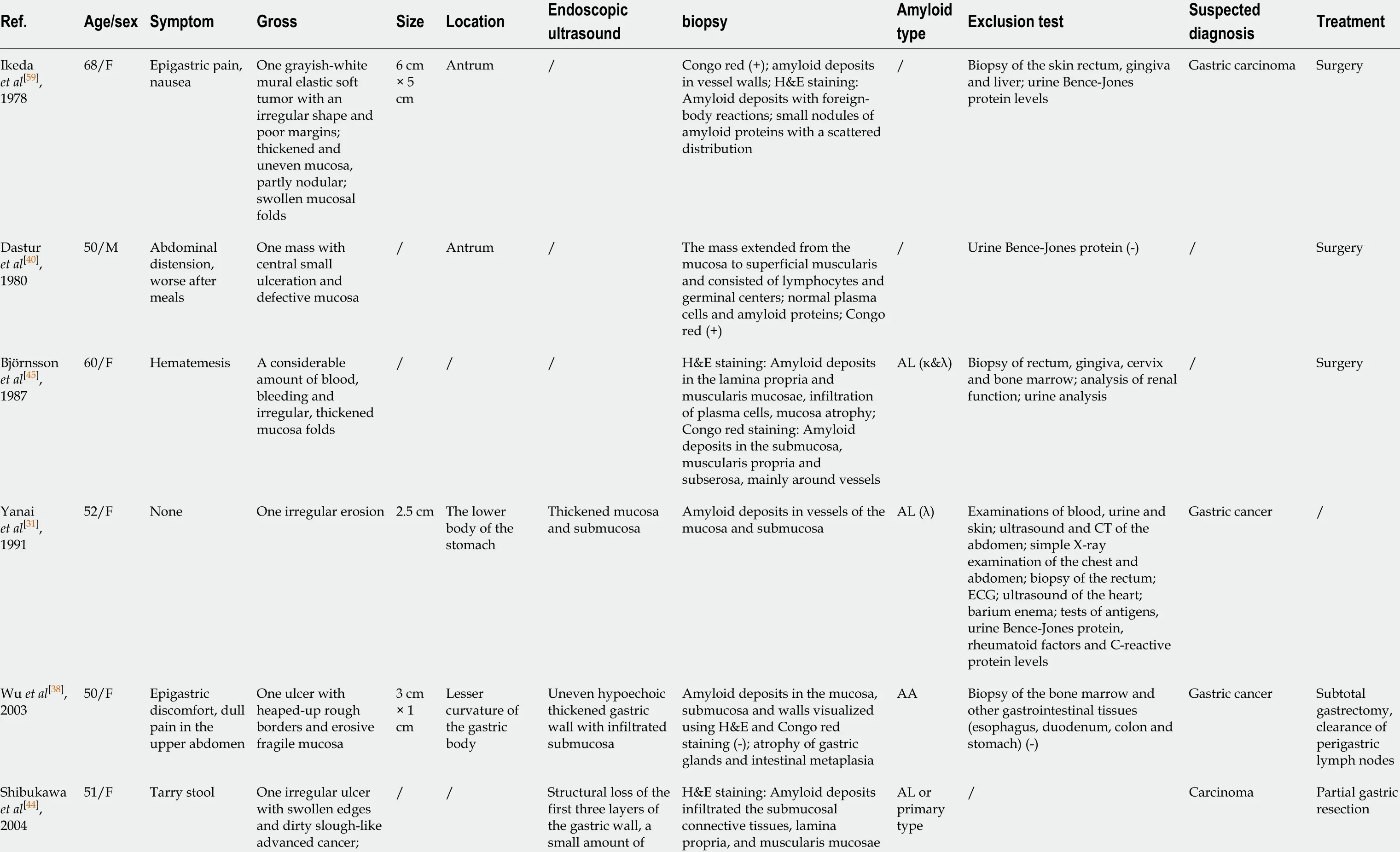
Table 1 Collection of recent case reports of localized gastric amyloidosis
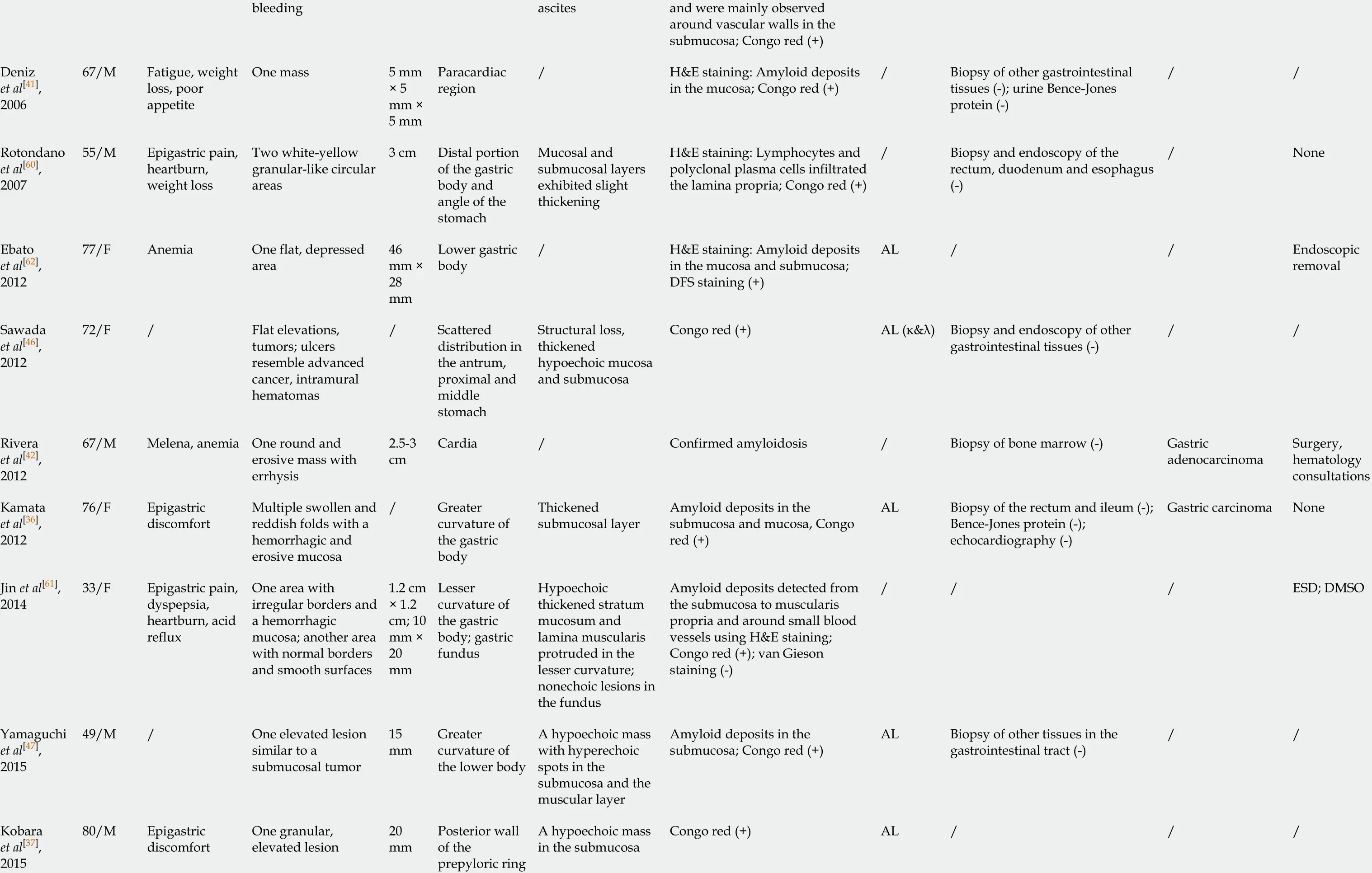
bleeding ascites and were mainly observed around vascular walls in the submucosa; Congo red (+)Deniz et al[4 1 ],200667 /M Fatigue, weight loss, poor appetite One mass 5 mm× 5 mm ×5 mm Paracardiac region/H&E staining: Amyloid deposits in the mucosa; Congo red (+)/Biopsy of other gastrointestinal tissues (-); urine Bence-Jones protein (-)//Rotondano et al[6 0 ],200755 /M Epigastric pain,heartburn,weight loss Two white-yellow granular-like circular areas 3 cm Distal portion of the gastric body and angle of the stomach Mucosal and submucosal layers exhibited slight thickening H&E staining: Lymphocytes and polyclonal plasma cells infiltrated the lamina propria; Congo red (+)/Biopsy and endoscopy of the rectum, duodenum and esophagus(-)/None Ebato et al[6 2 ],201277 /F Anemia One flat, depressed area 46 mm ×28 mm Lower gastric body/H&E staining: Amyloid deposits in the mucosa and submucosa;DFS staining (+)AL //Endoscopic removal Sawada et al[4 6 ],201272 /F /Flat elevations,tumors; ulcers resemble advanced cancer, intramural hematomas/Scattered distribution in the antrum,proximal and middle stomach Structural loss,thickened hypoechoic mucosa and submucosa Congo red (+)AL (κ&λ)Biopsy and endoscopy of other gastrointestinal tissues (-)//Rivera et al[4 2 ],201267 /M Melena, anemia One round and erosive mass with errhysis 2 .5 -3 cm Cardia /Confirmed amyloidosis /Biopsy of bone marrow (-)Gastric adenocarcinoma Surgery,hematology consultations Kamata et al[3 6 ],201276 /F Epigastric discomfort Multiple swollen and reddish folds with a hemorrhagic and erosive mucosa/Greater curvature of the gastric body Thickened submucosal layer Amyloid deposits in the submucosa and mucosa, Congo red (+)AL Biopsy of the rectum and ileum (-);Bence-Jones protein (-);echocardiography (-)Gastric carcinoma None Jin et al[6 1 ],201433 /F Epigastric pain,dyspepsia,heartburn, acid reflux One area with irregular borders and a hemorrhagic mucosa; another area with normal borders and smooth surfaces 1 .2 cm× 1 .2 cm; 10 mm ×20 mm Lesser curvature of the gastric body; gastric fundus Hypoechoic thickened stratum mucosum and lamina muscularis protruded in the lesser curvature;nonechoic lesions in the fundus Amyloid deposits detected from the submucosa to muscularis propria and around small blood vessels using H&E staining;Congo red (+); van Gieson staining (-)///ESD; DMSO Yamaguchi et al[4 7 ],201549 /M /One elevated lesion similar to a submucosal tumor 15 mm Greater curvature of the lower body A hypoechoic mass with hyperechoic spots in the submucosa and the muscular layer Amyloid deposits in the submucosa; Congo red (+)AL Biopsy of other tissues in the gastrointestinal tract (-)//Kobara et al[3 7 ],201580 /M Epigastric discomfort One granular,elevated lesion 20 mm Posterior wall of the prepyloric ring A hypoechoic mass in the submucosa Congo red (+)AL ///

ESD: Endoscopic submucosal dissection; DMSO: Dimethyl sulfoxide; CT: Computed tomography; ECG: Electrocardiograph; AL: Light chain; AA: Amyloid A; PFS: Progression-free survival.
After reviewing our data, a reasonable hypothesis is that amyloid subtypes and endoscopic findings are correlated, based on the similar appearance and features noted among the patients with the same amyloid subtype. The endoscopic appearance corresponded to amyloid subtypes in the small intestine, which is the most frequent site of gastrointestinal amyloidosis. For instance, the endoscopic presentation of AL is mostly thickening (75 %) and multiple polypoid protrusions (63 %), whereas a finegranular appearance is predominant in patients with AA (85 %)[49]. However, no association has been confirmed in the gastrointestinal tract according to a study with a small sample size, consistent with the results of the study by Samar M[39 ,50].Considering the small sample sizes and limitations of published studies, further studies with large samples are required.

Figure 4 Common symptoms described in case reports of localized gastric amyloidosis and the present times.
Narrow-band imaging with magnifying endoscopy (NBI-ME), a convenient technique that visualizes the mucosal morphology and microvascular architecture without a dye[51], is also highlighted in the amyloidosis diagnosis. Commonly, it is applied to detect malignancy in the colon and rectum through precise measurements of pit patterns with a promising accuracy (93 .4 %), sensitivity (100 %), and specificity(75 %)[52]. To date, the emerging use of NBI-ME has been reported not only in patients with LGA[32 ,46 ,47 ,53]but also in patients with amyloidosis of the rectum, trachea, and pleura[54 -56]. Upon enhancement, special pathological patterns are detected in affected lesions, manifesting as abnormal distorted vascular networks on the mucosa with a grayish-green appearance and grooved surface, which may relate to amyloid protein deposits[32 ,46 ,56]. The application of NBI-ME provides a better method to reveal the amyloid deposits as a useful tool to assist with the diagnosis.
Endoscopic ultrasound (EUS) is also useful for determining the diagnosis by revealing the loss of the normal structure and thickened hypoechoic gastric walls,which mainly affects the mucosa and submucosa (Table 1 ). Its use in combination with other methods has certain clinical value, such as EUS-guided fine needle aspiration(EUS-FNA). In a retrospective study of 47 patients with amyloidosis presenting with lymphadenopathy (swollen lymph nodes), the involvement of the gastrointestinal tract was noted in 39 % of cases[57]. In addition, EUS-FNA displayed a favorable sensitivity(83 %), specificity (94 %), and accuracy (86 %) for distinguishing malignancy, although the size of the swollen lymph nodes may influence the accuracy[58]. The clearance of peripheral lymph nodes was once suggested as a preferred method to prevent malignancy[38]. However, with the assistance of EUS-FNA, the resection of swollen lymph nodes is not necessary in every case. Further use of EUS is expected.
Biopsy
Biopsy is an essential assessment for diagnosing amyloidosis. Direct biopsy verification in patients with symptoms is the main diagnostic criterion for gastrointestinal amyloidosis, according to the guideline of AL amyloidosis[30]. Specific staining methods, such as H&E staining and Congo red staining, are used to visualize the amyloid deposits. Under a light microscope, amyloid deposits typically manifest as amorphous eosinophilic hyaline materials after H&E staining[38]. Amyloids with foreign-body reactions[43 ,59 ] and plasma cell infiltration[40 ,45 ,60]are also noted. The gold standard for identifying amyloid protein is Congo red staining. When bound to the dye, amyloid deposits are orange under a light microscope, and exhibit green, orange,or yellow birefringence under a polarized microscope[1,12]. Congo red staining is commonly used to diagnose all types of amyloidosis and has been applied to various tissue samples. Most of the case reports collected in the present study use and present the results of both of H&E and Congo red staining to confirm the diagnosis. Biopsy findings also show that amyloids are mainly deposited in the lamina propria,submucosa, and mucosa. Amyloid involvement of blood vessels is also observed[31 ,38 ,59 ,61]. AA LGA is characterized by deposits in the mucosa, submucosa, and vascular walls[38]. AL LGA deposits are mainly located in the mucosal propria,submucosa, and mucosa[32 ,34 ,36 ,47 ,62 ]. In a study of 79 cases of gastric amyloidosis,depositions occurred in the muscularis mucosae, but our data do not reveal this characteristic, likely due to the limited number of cases and exclusion of patients with systemic gastric amyloidosis[39]. Interestingly, the amyloid subtypes may account for the difference in deposit locations, which has been verified in several studies. In patients with gastric amyloidosis, AA amyloid deposits are preferentially located in the lamina propria, while AL is mainly deposited in the muscularis mucosae[63].
Although useful, Congo red staining has limitations. For example, when inadequate amyloid labeling occurs or the procedure is performed by inexperienced examiners in poorly controlled conditions, it presents limitations and improved methods have been constantly introduced over the years[14 ,64]. The application of some hypersensitive techniques may improve the accuracy of amyloid detection. Luminescent dyeconjugated polymer (LCP) spectroscopy detects every deposit that Congo red does using an easy method and effectively reduces false positives[65]. Based on these findings, several probes have been used in experiments. In a study with a small sample of patients with systemic amyloidosis, heptameric formic thiophene acetic acid(h-FTAA) was reported to be more sensitive than Congo red staining when detecting amyloid in abdominal fat[66]. Furthermore, h-FTTA detects small amyloid-like structures that are negative for Congo red staining due to its high quantum field. Thus,this method might potentially achieve the early diagnosis of amyloidosis and allow considerable progress in early treatment. Moreover, h-FTTA possesses a high sensitivity and relatively low specificity, but its combination with Mayer’s hematoxylin staining may compensate for its disadvantage in visual contrast[67].Although the methods that we mentioned above are still in development, a reasonable expectation is the future development of a hypersensitive approach as a replacement for Congo red staining.
Assessment of amyloid typing
Different forms of amyloid lead to completely different prognoses, and amyloid typing is a routine test performed in patients with amyloidosis. Several assessments have been applied for amyloid typing, including immunohistochemistry (IHC),immunofluorescence (IF) staining, immunoelectron microscopy, and genetic testing[8,16 ,39 ]. However, only half (13 /22 ) of the patients receive the test during the diagnosis of LGA, probably due to insufficient tissues and limited techniques[8]. IHC is currently used in the remaining patients and in patients with other forms of amyloidosis, as it is a convenient and rapid method. IHC was demonstrated to be a convincing approach with considerable specificity and sensitivity, according to a systematic study involving 117 patients[68]. However, some of its disadvantages include the presence of unclassifiable and misdiagnosed conditions due to the limited availability, specificity, and sensitivity of antibodies[16 ,65]. Laser microdissection/mass spectrometry (LMD/MS) requires a small amount of sample and exhibits a high specificity and sensitivity in typing clinical specimens[39 ,69]. LMD/MS has also been used to analyze samples that do not reach the standards for IF and exhibits a high detection rate of 92 % compared to 45 % using IHC[70 ,71]. Moreover, its use in combination with multiple reaction monitoring or liquid chromatography (LMD-LCMS) results in the detection of amyloid protein that IHC fails to detect and achieves early amyloid detection, even when Congo red results are negative[72 ,73]. In addition,the decellularization of an amyloid biopsy can facilitate the diagnosis of AL and ATTR amyloidosis, which are rich in plasma proteins. Here, decellularization provides a solution by removing the unnecessary proteins without altering the amyloid deposits and the basic structures of the biopsy[74 ,75]. Hopefully, these approaches may be used after further testing and reduce costs. Until then, we still call for the urgent increase in the use of IHC to diagnose LGA.
DIFFERENTIAL DIAGNOSIS
Systemic amyloidosis
The final diagnosis must be confirmed by the exclusion of the systemic involvement of other organs, which is generally performed using ultrasound of the heart and kidney and biopsy of the bone marrow and regions of the gastrointestinal tract, including the esophagus, duodenum, and colon. Occasionally, urine levels of the Bence-Jones protein and other antigens are tested. Under most conditions, the aforementioned tests are selected based on clinicians’ experience with a screening procedure. Immunohistochemical examinations also provide a lead for the differentiation of systemic and localized amyloidosis, because the deposited protein exhibits specific distribution patterns. For example, AA and ATTR are often detected in the former type, while AL has been detected in both types[2,11].
Some radiopharmaceuticals present potential abilities to distinguish systemic and localized amyloidosis. SAP is one of the nonfibrillar components present in all types of amyloid deposits, and its abundant accumulation makes it an ideal radiotracer to visualize amyloid deposits in images of the body[2,22 ]. In a study of 189 patients with confirmed amyloidosis,123 I SAP scintigraphy, a noninvasive qualitative method,presented a high sensitivity and specificity and was applied to diagnose most cases of AA and AL amyloidosis[76]. Through the use of whole-body scintigraphy, the injection of123 I SAP obviously reveals the distribution and extent of amyloid deposits in images that a histopathological examination fails to detect, and the organ involvement identified using this technique exceed the results obtained in the clinic, which may be valuable in excluding systemic involvement. The only limitation is its failure in revealing the heart muscle[76]. It is gradually becoming a universal technique used in relevant studies for scanning systemic involvement in the whole body, except for the heart[3,77]. With the development of proper radiotracers and a considerable increase in use, we postulate that nuclear images will significantly contribute to the diagnosis of LGA.
18 F-fluorodeocyglucose positron emission tomography/computed tomography (18 FFDG PET/CT) is also reported as a potential technique to distinguish systemic and localized amyloidosis. Under most conditions, it is introduced as a method for detecting lung malignancy in patients suspected of having the disease, and it is mainly used to scan amyloidosis in the lung[78 ,79 ]. Glaudemans et al[77 ] reported that 18 F-FDG PET/CT is able to visualize the difference between localized and systemic amyloidosis by imaging the inflammatory reaction of multinuclear giant cells, which are unique in localized amyloidosis, manifesting as positive FDG uptake at sites of amyloid deposits in patients with localized amyloidosis but negative uptake in patients with systemic amyloidosis[77 ]. However, Mekinian et al[80]also reported positive results in patients with systemic amyloidosis, which may be attributed to the limited number of samples or inappropriate designs[80 ]. To date, 18 F-FDG PET/CT is still an immature method to exclude systemic amyloidosis, and patients with gastric amyloidosis were not examined in either study. When used in conjunction with confirmed evidence obtained from other techniques, it may play a supporting role in determining the diagnosis.
Advanced cancer
Due to its rarity and nonspecific presentations, amyloidosis is typically not the first diagnosis suspected by clinicians, and differential diagnosis becomes an important step. Under most circumstances, patients are scheduled for a precise test screening for potential cancers and are confirmed to have amyloidosis. Most lesions were suspected to be advanced gastric cancers or some gastrointestinal tumors (9 /22 ) since they share common appearances, such as ulcers, elevations, and tumor-like lesions. During gastroscopy or esophagogastroduodenoscopy, some of these lesions are easily suspected to be submucosal tumors[34 ,46]. The most reliable method for excluding the possibility of tumors is a tissue biopsy that does not detect tumor components and the confirmation of the presence of amyloid based on positive Congo red staining results.Tumor biomarkers are occasionally involved in the examinations based on clinicians’experience. Although NBI-ME is a new technique, it may also facilitate differentiation,given its ability to exclude malignancies. Based on the clear visualization of the microvasculature and microstructure under NBI-ME, vascular and surface pattern classifications are proposed as a reference, and the typical hallmarks of advanced gastric cancer can be observed, thus providing a reliable evaluation of advanced gastric cancer[81]. A subsequent study reported an obviously increased sensitivity and accuracy of NBI-ME compared with routine approaches in scanning for advanced gastric cancer, and the use of NBI-ME was also beneficial to locate the most suspicious lesion for biopsy[82].
Compared to other case reports, we excluded advanced cancer using a more innovative screening method, namely, immunohistochemical staining, given our restricted conditions. Because gastric carcinoma is suspected in some cases[31 ,36 ,59], it may be a wise choice. The combination of several antibodies for immunohistochemical staining, including CKPAN, KL067 , Periodic Acid-Schiff, spinal muscular atrophy and vimentin, with biopsy results did not reveal strong support for cancers and excluded the possibility of gastric cancer from various origins.
TREATMENT AND PROGNOSIS
Generally, the goal of therapy for amyloidosis is to eliminate harmful extracellular amyloid deposits and restore the normal function of affected organs as much as possible. The main treatments include surgery, observation, and radiotherapy[83].Chemotherapy is recommended for patients with AL who present with myeloma.
Several novel techniques are also introduced here. As we mentioned above, SAP is a type of plasma protein present in all amyloid deposits, and its interaction with amyloid fibrils may prevent the digestion of amyloid, as evidenced by the results of in vitro experiments[21 ,22]. Because amyloidosis is delayed in SAP knockout mice, SAP clearance is introduced as a potential strategy for treating amyloidosis with the application of relevant antibodies and drugs[24 ]. Here, (R)-1 -{6 -[(R)-2 -carboxypyrrolidin-1 -yl]-6 -oxo-hexanoyl}pyrrolidine-2 -carboxylic acid (CPHPC), a smallmolecule drug with a high affinity for SAP, depletes most of the circulating SAP[84 ,85].Its use in combination with the anti-SAP antibody dezamizumab results in a considerable reduction in the amount of residual amyloid protein visible in123 I SAP scintigraphy, with no obvious side effects[85 ,86]. After treatment, patients tend to present an improved or stable state, suggesting its potential ability to ameliorate symptoms and reduce damage in the affected organs[84]. A subsequent study further confirmed its effects on the spleen, kidney, and liver[87]. Currently, studies targeting SAP are mainly conducted in mice and small samples of patients with systemic amyloidosis. Although complete removal of all amyloid deposits has not yet been achieved, its usage in patients with LGA as a rapid method for the early clearance of amyloid deposits is worthy of further exploration.
Currently, the first-line therapy for localized AL gastrointestinal amyloidosis is mainly observation/supportive care and the excision of amyloid deposits, and radiotherapy is rarely used, according to the experience of physicians from the Mayo Clinic[83 ]. Among 13 patients with LGA presented with clear therapeutic strategies,surgery was the main choice (8 /13 ), and a few patients chose observation (4 /13 ). The administration of dimethyl sulfoxide is also recommended to reduce the digestive symptoms and result in a visible improvement on endoscopy[61].
The prognosis of amyloidosis is generally related to the extent of organ damage.Unlike systemic amyloidosis, localized amyloidosis has an excellent prognosis and minimally affects patient survival[3]. After first-line treatment, most patients improved(53 %) or achieved a stable state (31 %), and only a few progressed (0 .2 %), according to a study enrolling 413 patients with localized AL amyloidosis. However, the study also mentioned an undeniable recurrence rate, as two or more recurrences occurred in 5 %of the 413 patients, and the first 5 years after diagnosis is a crucial period for recurrence[83]. To date, no recurrence of LGA has been mentioned in the case reports published, and all patients presented a healthy state in follow-up visits, but close follow-up and regular examinations are necessary, particularly within the first 5 years(Figure 6 ).
CONCLUSION
Local gastric amyloidosis is such an extremely rare disease that only 22 cases have been reported in the past few decades and the disease is unknown to the public. It is commonly introduced as a human pathological state in which an abnormally misfolded protein accumulates in tissues, causing structure loss, organ dysfunction,and even death. Its pathogenesis remains a mystery, but a few influencing factors that may contribute to the formation of amyloid fibrils are studied and introduced here.Our review may inspire further investigations of the mechanism. Based on the 21 existing cases and our case, we present a detailed description of the main information available on LGA and conclusions regarding its clinical features, diagnostic tools, and treatment, with the goal of establishing future guidelines. Its clinical manifestations are complex and similar to those of other gastric diseases, such as advanced cancer,resulting in minimal awareness among clinicians. The diagnostic tools include biopsy,imaging, and amyloid typing. The final diagnosis mainly depends on the biopsy results, and Congo red staining remains the gold standard. Treatments for LGA mainly include supportive care and surgery. After treatment, most patients receive a good prognosis. Currently, due to its rare incidence, LGA lacks public awareness, and studies that explore its pathogenesis and its clinical features are often unspecific. For clinicians, LGA is a challenge to diagnose using regular tests. Building on that information, we describe the main clinical features and take the lead in proposing a process for diagnosing LGA from the clinicians’ perspective, with the aims of promoting awareness of LGA and potentially contributing to the development of LGA guidelines.
 World Journal of Gastroenterology2021年12期
World Journal of Gastroenterology2021年12期
- World Journal of Gastroenterology的其它文章
- Cascade of care for children and adolescents with chronic hepatitis C
- R2 * value derived from multi-echo Dixon technique can aid discrimination between benign and malignant focal liver lesions
- Risk perception and knowledge of COVID-19 in patients with celiac disease
- Risk stratification and geographical mapping of Brazilian inflammatory bowel disease patients during the COVID-19 outbreak: Results from a nationwide survey
- Hepatitis E in solid organ transplant recipients: A systematic review and meta-analysis
- Emerging wearable technology applications in gastroenterology: A review of the literature