
Synthesis,Structure and Catalytic Properties of Mn(Ⅱ)Coordination Polymer through in Situ Ligand Reaction
2021-04-10 14:20:12ZHAOSuQinGUJinZhong
无机化学学报 2021年4期
ZHAO Su-Qin GU Jin-Zhong
(1 College of Physics and Electronic Information Engineering,Qinghai University for Nationalities,Xining 810007,China)
(2 College of Chemistry and Chemical Engineering,Lanzhou University,Lanzhou 730000,China)
Abstract:One 2D coordination polymer,namely[Mn(μ3-Hdpna)(2,2′-bipy)]n(1),has been constructed hydrothermally using Hdbna(Hdbna=5-(2,3-dicyanobenzoxy)nicotic acid),2,2′-bipy(2,2′-bipy=2,2′-bipyridine),and manganese chloride at 160℃.Interestingly,the H3dpna(H3dpna=5-(2,3-dicarboxylphenoxy)nicotic acid)ligand was generated by in situ hydrolysis of cyano groups in Hdbna.The product was isolated as stable crystalline solid and was characterized by IR spectrum,elemental analysis,thermogravimetric analysis(TGA),and single-crystal X-ray diffraction analysis.Single-crystal X-ray diffraction analysis reveals that compound 1 crystallizes in the triclinic system,space group P.Compound 1 discloses a 2D sheet with an hcb topology.Compound 1 was applied as an efficient heterogeneous catalyst for the cyanosilylation.CCDC:2025016.
Keywords:coordination polymer;ether-bridged tricarboxylic acid;catalytic properties;in situ reaction
0 Introduction
In recent years,the design and hydro(solvo)thermal in situ syntheses of metal-organic coordination plymers have attracted great interest in the field of coordination chemistry and organic chemistry not only because of their intriguing architectures and topologies,but also for that they have shown a variety of potential applications in catalysis,magnetism,luminescence and gas absorption[1-8].Commpared with the traditional synthesis method,hydrothermal and solvothermal method could create more chances for in situ syn-these of ligands due to the reaction conditions of high temperature and high pressure[9-12].At the same time,the hydro(solvo)thermal method has demonstrated increasing success in providing alternative pathways to crystalline complexes with in situ synthesized ligands which are difficult to obtain by routine synthetic methods.
Following our interest in the exploration of novel and poorly investigated multicarboxylic acids for the design of coordination polymers[13-17],recently,we began to construct coordination polymers by use of the advantage of in situ ligand reaction.On the basis of current research on in situ ligand reaction,the carboxylatebased ligands can be in situ generated from the CN-containing ligands precursor[7,18-19].Thus,the precursor we selected is 5-(2,3-dicyanobenzoxy)nicotic acid(Hdbna)due to its following characteristics:(1)it contains one carboxylate and two CN groups,which can form a tricarboxylate ligand through in situ ligand reaction(Scheme 1);(2)its corresponding acid,5-(2,3-dicarboxylphenoxy)nicotic acid(H3dpna),still remain largely unexplored in the construction of coordination polymers[20].
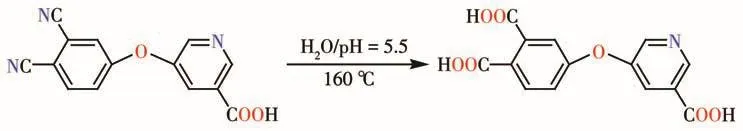
Scheme 1 H3 dpna ligand formed via in situ reaction
Herein,we report the synthesis,crystal structure,and catalytic properties of Mn(Ⅱ)coordination polymer with H3dpna ligands.
1 Experimental
1.1 Reagents and physical measurements
All chemicals and solvents were of AR grade and used without further purification.Carbon,hydrogen and nitrogen were determined using an Elementar Vario EL elemental analyzer.IR spectrum was recorded using KBr pellets and a Bruker EQUINOX 55 spectrometer.Thermogravimetric analysis(TGA)data were collected on a LINSEIS STA PT1600 thermal analyzer with a heating rate of 10℃·min-1.Powder X-ray diffraction(PXRD)patterns were measured on a Rigaku-Dmax 2400 diffractometer using Cu Kα radiation(λ=0.154 06 nm);the X-ray tube was operated at 40 kV and 40 mA.The data collection range was between 5°and 45°.Solution1H NMR spectra were recorded on a JNM ECS 400M spectrometer.
1.2 Synthesis of[Mn(μ3-Hdpna)(2,2′-bipy)]n(1)
A mixture of MnCl2·4H2O(0.040 g,0.2 mmol),Hdbna(0.053 g,0.2 mmol),2,2′-bipy(0.031 g,0.2 mmol),NaOH(0.016 g,0.4 mmol),and H2O(10 mL)was stirred at room temperature for 15 min,and then sealed in a 25 mL Teflon-lined stainless steel vessel,and heated at 160℃for 3 d,followed by cooling to room temperature at a rate of 10℃·h-1.Yellow blockshaped crystals were isolated manually,and washed with distilled water.Yield:55%(based on Hdbna).Anal.Calcd.for C24H15MnN3O7(%):C 56.26,H 2.95,N 8.20;Found(%):C 56.48,H 2.93,N 8.16.IR(KBr,cm-1):1 688w,1 598m,1 553s,1 470s,1 437s,1 412m,1 371m,1 301w,1 256m,1 210w,1 148w,1 062w,1 017w,976w,905w,844w,819w,794w,761m,736w,716w,695w,629w.
The compound is insoluble in water and common organic solvents,such as methanol,ethanol,acetone and DMF.
1.3 Structure determination
The single crystal with dimensions of 0.24 mm×0.21 mm×0.20 mm was collected at 293(2)K on a Bruker SMART APEX Ⅱ CCD diffractometer with Cu Kα radiation(λ =0.154 178 nm).The structure was solved by direct methods and refined by full matrix least-square on F2using the SHELXTL-2014 program[21].All non-hydrogen atoms were refined anisotropically.All the hydrogen atoms were positioned geometrically and refined using a riding model.A summary of the crystallography data and structure refinement for 1 is given in Table 1.The selected bond lengths and angles for compound 1 are listed in Table 2.Hydrogen bond parameters of compound 1 are given in Table 3.

Table 1 Crystal data for compound 1
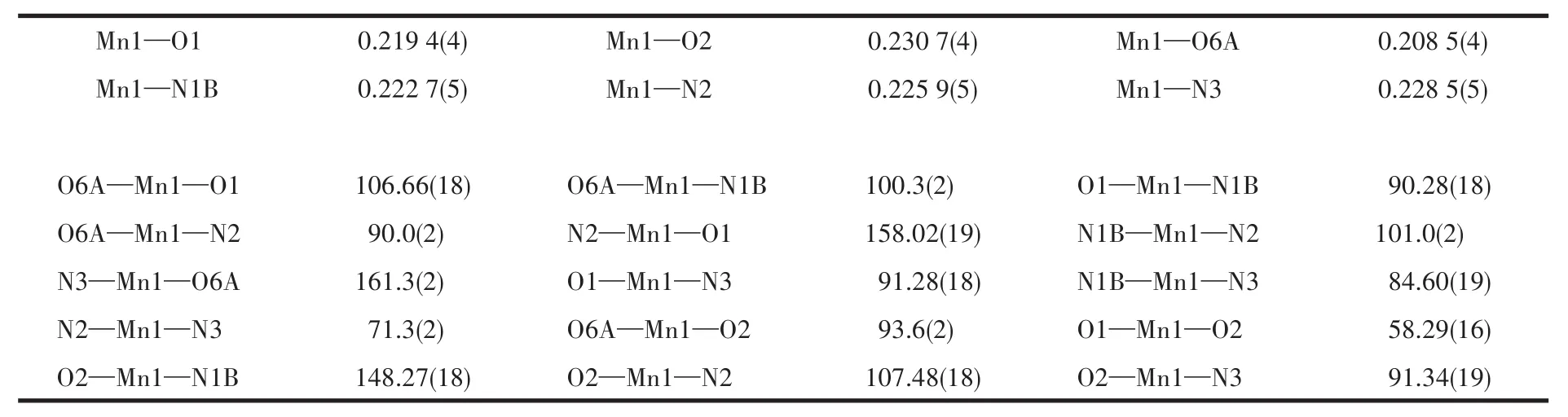
Table 2 Selected bond distances(nm)and bond angles(°)for compound 1

Table 3 Hydrogen bond parameters of compound 1
CCDC:2025016.
1.4 Catalytic cyanosilylation of aldehydes
In a typical test,a suspension of an aromatic aldehyde(0.50 mmol,4-nitrobenzaldehyde as a model substrate),trimethylsilyl cyanide(1.0 mmol)and catalyst(molar fraction:3%)in dichloromethane(2.5 mL)was stirred at room temperature.After a desired reaction time,the catalyst was removed by centrifugation,followed by an evaporation of the solvent from the filtrate under reduced pressure to give a crude solid.This solid was dissolved in CDCl3and analyzed by1H NMR spectroscopy for quantification of products(Fig.S1,Supporting information).To perform the recycling experiment,the catalyst was isolated by centrifugation,washed with dichloromethane,dried at room temperature,and reused.The subsequent steps were performed as described above.
2 Results and discussion
2.1 Description of the structure
X-ray crystallography analysis reveals that compound 1 crystallizes in the triclinic system space group P.As shown in Fig.1,the asymmetric unit of 1 bears one crystallographically unique Mn (Ⅱ) atom,one μ3-Hdpna2-block,and one 2,2′-bipy.The Mn1 center is six-coordinated and forms a distorted octahedral{MnN3O3}geometry.It is completed by three carboxylate O and one N atoms from three μ3-Hdpna2-blocks and two N atoms from the 2,2′-bipy moiety.The Mn—O and Mn—N bond distances are 0.208 5(4)~0.230 7(4)nm and 0.222 7(5)~0.228 5(5)nm,respectively;these are within the normal ranges observed in related Mn(Ⅱ)compounds[15-17].In 1,the Hdpna2-ligand adopts the coordination mode Ⅰ(Scheme 2)with two deprontonat-ed carboxylate groups being monodentate or bidentate.In the Hdpna2-ligand,a dihedral angle(between two aromatic rings)and a C—Oether—C angle are 69.75°and 116.43°,respectively.The μ3-Hdpna2-ligands connect Mn1 atoms to give a 2D sheet(Fig.2).This sheet is composed of the 3-connected,topologically equivalent Mn1 and μ3-Hdpna2-nodes(Fig.3).The resulting net can be described as a uninodal 3-connected layer with an hcb(Shubnikov hexagonal plane net/(6,3))topology and point symbol of(63).
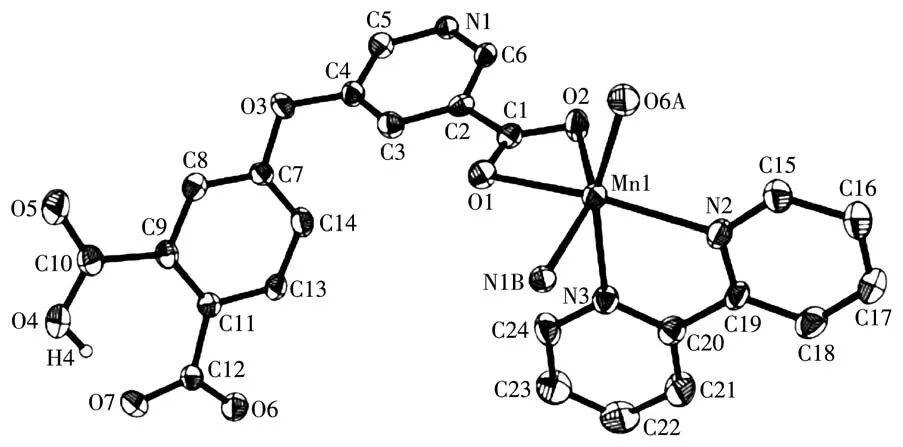
Fig.1 Drawing of asymmetric unit of compound 1 with 30% probability thermal ellipsoids

Scheme 2 Coordination mode of Hdpna2-ligand in compound 1
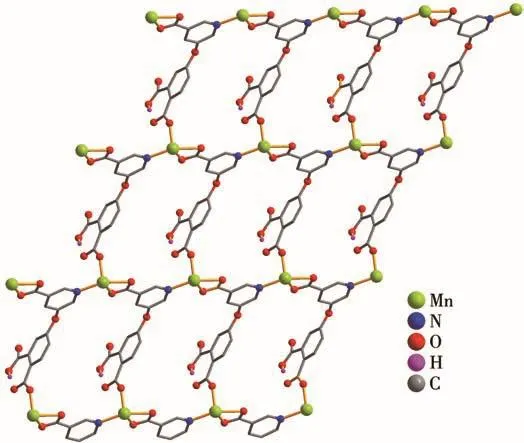
Fig.2 Perspective of 2D sheet along a and b axes in 1
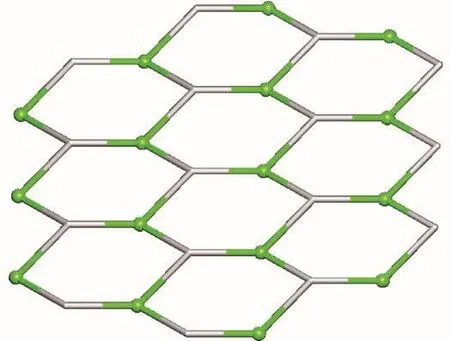
Fig.3 Topological representation of uninodal 3-connected metal-organic layer with hcb(Shubnikov hexagonal plane net/(6,3))topology viewed along c axis
2.2 TGA analysis
To determine the thermal stability of compound 1,its thermal behavior was investigated under nitrogen atmosphere by TGA.As shown in Fig.4,compound 1 did not contain solvent of crystallization or H2O ligands and remained stable up to 312℃,followed by a decomposition on further heating.

Fig.4 TGA curve of compound 1
2.3 Catalytic cyanosilylation of aldehydes
Given the potential of manganes(Ⅱ)coordination compounds to catalyze the organic reactions[22-23],we explored the application of 1 as a heterogeneous catalyst in the cyanosilylation of 4-nitrobenzaldehyde as a model substrate to give 2-(4-nitrophenyl)-2-((trimethylsilyl)oxy)acetonitrile.Typical tests were carried out by reacting a mixture of 4-nitrobenzaldehyde,trimethylsilyl cyanide(TMSCN)and a Mn catalyst in dichloro-methane at room temperature(Scheme 3,Table 4).Such effects as reaction time,catalyst loading,solvent composition,catalyst recycling and finally substrate scope were investigated.

Scheme 3 Mn-catalyzed cyanosilylation of 4-nitrobenzaldehyde(model substrate)

Scheme 4 Mechanism for the Mn-catalyzed cyanosilylation reaction
Upon using compound 1 as the catalyst(molar fraction:3%),a high conversion of 82% of 4-nitrobenzaldehyde into 2-(4-nitrophenyl)-2-((trimethylsilyl)oxy)acetonitrile was reached after 12 h in dichloromethane at room temperature(Table 4,Entry 7).Upon extending the reaction time further to 18 h,only a slight increase in the product yield to 83% occurred.Moreover,no other products were detected,and the yield of this product was considered the conversion of 4-nitrobenzaldehyde(Fig.S1).
We also compared the activities of catalyst 1 in the reactions of other substituted aromatic and aliphatic aldehydes with trimethylsilyl cyanide,and the corresponding cyanohydrin derivatives were produced in yields ranging from 43% to 82%(Table 5).Aryl aldehydes bearing strong electron-withdrawing substituents(e.g.,nitro and chloro)exhibited the higher reactivities(Table 5,Entry 2~5),which may be related to an increase in the electrophilicity of the substrate.Aldehydes containing electron-donating proups(e.g.,methyl)showed lower reaction yields(Table 5,Entry 7),as expected.An ortho-substituted aldehyde showed lower reactivity,possibly as a result of steric hindrance.
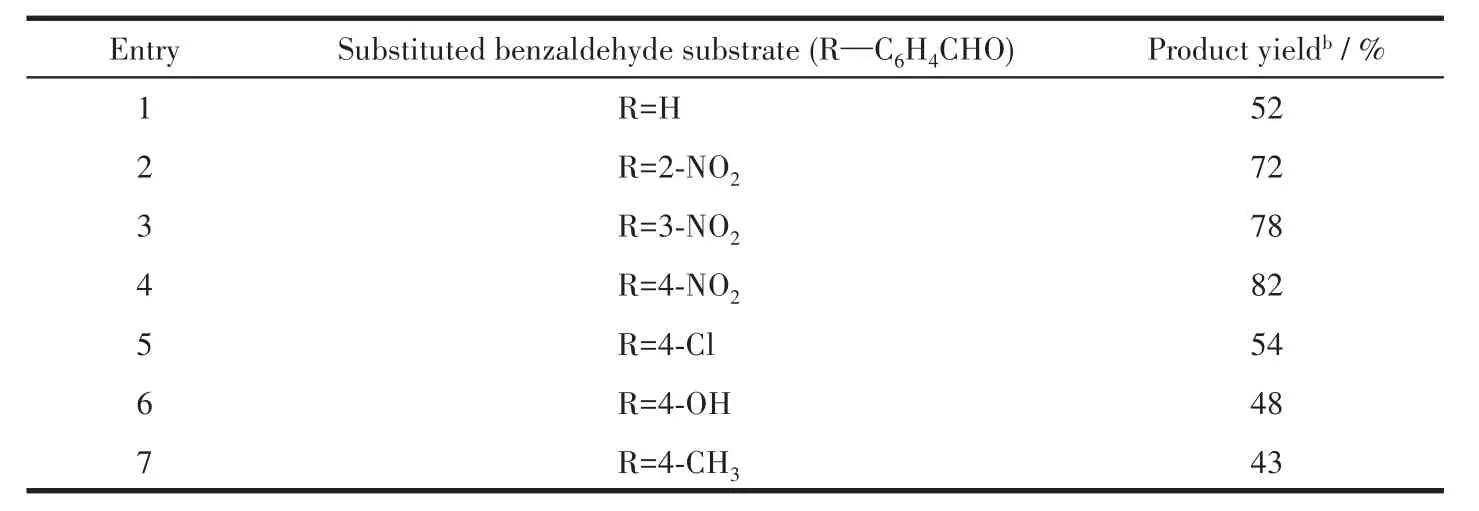
Table 5 Cyanosilylation of various aldehydes with TMSCN catalyzed by compound 1a
To examine the stability of 1 in the cyanosilylation reaction,we tested the recyclability of this heterogeneous catalyst.For this purpose,upon completion of a reaction cycle,we separated the catalyst by centrifugation,washed it with CH2Cl2,and dried it at room temperature before its further use.The catalytic system mained the high activity in at least five consecutive cycles(the yields were 80%,80%,79% and 78% for second to fifth run,respectively).According to the PXRD data(Fig.5).the structure of 1 was essentially preserved after five catalytic cycles.

Fig.5 PXRD patterns for 1
A possible catalytic cycle for the cyanosilylation reaction catalyzed by a Mn coordination polymer is proposed in Scheme 4.It can involve dual activation of the carbonyl and TMSCN by the Mn(Ⅱ)centre and a ligated carboxylate group,respectively,followed by the formation of the C—C bond leading to cyanohydrin[24-25].
3 Conclusions
In summary,we have synthesized one Mn(Ⅱ)coordination polymer 1 based on a tricarboxylate ligand generated by in situ reaction.Compound 1 shows a 2D sheet structure,and it exhibits a higher catalytic activity in the cyanosilylation at room temperature.
Supporting information is available at http://www.wjhxxb.cn

- 无机化学学报的其它文章
- Lamellar-Smectic Polyoxometalate Liquid Crystal Encapsulated by Branched Bola-Form Amphiphiles
- Synthesis,Structure,Magnetic and Photocatalytic Properties of Nickel(Ⅱ)Coordination Polymer Based on 1-(3,5-Dicarboxybenzyl)-1H-pyrazole-3,5-dicarboxylic Acid Ligand
- Flexible Bis(imidazole)-Based Ligands Oriented Co(Ⅱ)-Glutarate Metal-Organic Frameworks:Syntheses,Structures and Properties
- 以四乙基氢氧化铵为模板并通过转晶法合成高硅铝比的SSZ-13沸石分子筛
- Fe物种改性海胆状Nb2O5光催化降解类吩噻嗪染料
- Cu3P纳米板阵列@泡沫铜三维骨架的原位构筑及其作为高性能锂金属负极