
RePhine:An Integrative Method for Identification of Drug Response-related Transcriptional Regulators
2021-03-30 02:46XujunWangZhengtaoZhangWenyiQinShiyiLiuCongLiuGeorgiGenchevLijianHuiHongyuZhaoHuiLu
Xujun Wang ,Zhengtao Zhang ,Wenyi Qin ,Shiyi Liu ,Cong Liu ,Georgi Z.Genchev,6 ,Lijian Hui,Hongyu Zhao,7,*,Hui Lu,3,7,8,9,*
1SJTU-Yale Joint Center for Biostatistics and Data Science,Department of Bioinformatics and Biostatistics,School of Life Science and Biotechnology,Shanghai Jiao Tong University,Shanghai 200240,China
2State Key Laboratory of Cell Biology,CAS Center for Excellence in Molecular Cell Science,Shanghai Institute of Biochemistry and Cell Biology,Shanghai 200031,China
3Department of Bioengineering,University of Illinois at Chicago,Chicago,IL 60607,USA
4Department of Genetics,School of Medicine,Yale University,New Haven,CT 06511,USA
5Department of Medical Informatics,Columbia University,New York,NY 10032,USA
6Bulgarian Institute for Genomics and Precision Medicine,Sofia 1000,Bulgaria
7Department of Biostatistics,Yale School of Public Health,New Haven,CT 06511,USA
8Institute of Science and Technology for Brain-Inspired Intelligence,Fudan University,Shanghai 200433,China
9Center for Biomedical informatics,Shanghai Children’s Hospital,Shanghai 200040,China
Abstract Transcriptional regulators (TRs) participate in essential processes in cancer pathogenesis and are critical therapeutic targets.Identification of drug response-related TRs from cell line-based compound screening data is often challenging due to low mRNA abundance of TRs,protein modifications,and other confounders (CFs).In this study,we developed a regression-based pharmacogenomic and ChIP-seq data integration method (RePhine) to infer the impact of TRs on drug response through integrative analyses of pharmacogenomic and ChIP-seq data. RePhine was evaluated in simulation and pharmacogenomic data and was applied to pan-cancer datasets with the goal of biological discovery.In simulation data with added noises or CFs and in pharmacogenomic data,RePhine demonstrated an improved performance in comparison with three commonly used methods(including Pearson correlation analysis,logistic regression model,and gene set enrichment analysis).Utilizing RePhine and Cancer Cell Line Encyclopedia data,we observed that RePhinederived TR signatures could effectively cluster drugs with different mechanisms of action.RePhine predicted that loss-offunction of EZH2/PRC2 reduces cancer cell sensitivity toward the BRAF inhibitor PLX4720.Experimental validation confirmed that pharmacological EZH2 inhibition increases the resistance of cancer cells to PLX4720 treatment.Our results support that RePhine is a useful tool for inferring drug response-related TRs and for potential therapeutic applications.The source code for RePhine is freely available at https://github.com/coexps/RePhine.
KEYWORDS Pharmacogenomics;ChIP-seq;Transcriptional regulator;BRAF inhibitor resistance;Drug resistance
Introduction
Cancer,one of the most common causes of death [1],is characterized by uncontrolled cell division [2,3].Pharmacological treatments such as chemotherapy,targeted therapy,and immunotherapy are widely used.Accurate drug selection has the potential to improve patient outcomes by matching the patient’s genomic characteristics with the most effective treatment available [4].The comprehensive delineation of the association between drug response and omics features by using a suitable computational model may significantly contribute to preclinical research and drive clinical decision-making.Most existing drug response prediction methods are based on direct putative correlations between individual genes’ mRNA levels and drug sensitivity measurement [4].Such predictions,however,are not always sufficiently robust due to the inherent experimental noises [5,6].The robust identification of drug response biomarkers remains a significant challenge.
Many transcriptional regulators (TRs),including transcription factors(TFs)and chromatin regulators(CRs),can control cell development and cell survival by regulating the expression of target genes [7-9].For example,inhibiting the TFs ESR1 and SP1 could induce the death of myeloma cells[10]and targeting the CR BRD4 rescues the response to the γ-secretase inhibitors in T-cell acute lymphoblastic leukemia [11].These studies reveal the complicated and vital roles of TRs and emphasize the necessity of systematic identification of drug response-related TRs.However,a number of biological features of TRs (e.g.,low mRNA abundance,protein modifications,and localizations) pose challenges to such identification [12-14].For example,by using Cancer Cell Line Encyclopedia (CCLE) data,the resistance role of zinc finger E-box binding homeobox 1(ZEB1)can be directly reflected by its mRNA level correlated to the response to the drug erlotinib,but there is no obvious correlation in the case of Fos proto-oncogene(FOS) (Figure S1A and B),though both TRs are known biomarkers of erlotinib[15,16].Therefore,solely relying on the mRNA levels of TRs to identify TR-drug response relationship lacks identification power (false negatives) and may even lead to inaccurate associations(false positives)in the existing compound screening data.Given the importance of TRs in tumorigenesis and the limitations of existing data,a new strategy is required to explore the functional roles of TRs in drug response[17].However,the effects of copy number variations(CNVs),data noises,and confounding mutations of some kinase genes or tumor suppressor genes,as well as how to accurately infer targets,pose challenges in this identification effort.
In this study,we hypothesized that the association of a TR with drug response could also be inferred through the association between the expression of its downstream targets and the drug treatment(Figure 1A).Based on this notion,we developed the regression-based pharmacogenomic and ChIP-seq data integration method (RePhine)which takes pharmacogenomic and ChIP-seq data as input and identifies TRs associated with response to pharmacological drug therapy(Figure 1B).We demonstrated thatRePhinehas an improved performance in comparison with several commonly used methods in TR identification in simulation and pharmacogenomic data.Through theRePhineanalysis pipeline,we characterized TR response signatures of drug treatments.UtilizingRePhine,we uncovered a BRAF inhibitor resistance mechanism relevant to the functional loss of polycomb repressive complex 2(PRC2).This mechanism was then validated by drug treatment experiments in cell lines and bioinformatic analyses in CRISPR screening data,The Cancer Genome Atlas(TCGA)patient cohorts,and differentially expressed genes between BRAF inhibitor-sensitive and resistant cells.
Method
Description of the RePhine method
TheRePhinemethod is designed to identify the TRs whose targets have concordant correlations with drug response(Figure 1B and C).RePhineadjusts for confounders (CFs)by calculating the partial correlation coefficients between expression of all the genes and one specific drug treatment across all the cell lines.Next,RePhinemeasures the TR targets quantitatively from ChIP-seq data.Then,RePhineidentifies the drug response-related TRs by regressing the TR target measurements on the partial correlation coefficients.RePhineemploys the two metrics — univariatePvalue (uniP) and multivariatePvalue (multiP) — to evaluate the significance of the associations of TRs to the given drug.
TheRePhinemethod consists of the four steps that were described as below(Figure 1C,Figure S2;File S1).

Figure 1 A schematic view of RePhine hypothesis and analysis workflow
Step 1:CF identification and correction
In Step 1.1,the effect of CNVs is adjusted.To account for the fact that gene copy numbers can influence gene expression levels independent of TR regulation[18],RePhineutilizes a linear model to evaluate the impact of a TR on the expression of its targets.
The following model is fitted:

whereis the copy number of a certain gene in the cell linen;is the expression value of this gene in the same cell line;the fitted residualis considered as the adjusted expression;is the coefficient.
In step 1.2,mutation CFs are identified.Some somatic mutations can influence drug response independent of TR regulation,and such mutations may confound the identification of drug response-related TRs.To identify such mutations related to drug response,a two-stage procedure is applied.
In step 1.2.1,genes with mutations at extremely low frequency across the cancer cell lines(<5%of the number of the cell lines) are filtered.
In step 1.2.2,Adaptive Lasso(AL)is then used to select significantly drug response-related mutated genes[19]:

The R “glmnet” package is utilized for step 1.2.2.A likelihood ratio test is used to estimate the significance ofβ.Genes with both significant uniPand multiP(P<0.01)are used as the CFs in the subsequent partial correlation computation step 1.3.
In step 1.3,the partial correlation coefficients are calculated to adjust the effect of the CFs(estimated from step 1.2) and accurately measure the association between the drug response and the expression of all the genes(estimated from step 1.1).The detailed calculation of the partial correlation coefficients is described in File S1.The R“ppcor”package is used to perform the computation for each gene in this step.
In step 1.4,the partial correlation coefficients of cancer type-specific genes are down weighted.Expression in tumorvs.normal samples in 14 cancer types,sourced from TCGA data,is compared before this step.ThenRePhineperforms down-weighting of the genes which are differentially expressed [calculated by“limma” package with false discovery rate(FDR)<0.001]in less than 1/3 of the cancer types.Then,RePhineis applied to a pan-cancer dataset such as CCLE,aiming to identify pan-cancer drug responserelated TRs.The goal of this step is to reduce the effect of cancer type-specific genes.For such genes,the partial correlation coefficients are set to zero.In our calculation,only 10% of the genes are corrected in this step and most TRs are not affected (Table S1).Compared to the nonfiltering procedure,the filtering step up-weights the TRs related to the common cancer pathways as expected(Table S1).This step is optional and it can be skipped when applyingRePhineto other datasets.
Step 2:regulatory potential calculation
To determine the targets of TRs from ChIP-seq data,we implement a modified model [20] by additionally considering peak signal strength;in the original model,only peak counts and distances to genes were considered.Regulatory potential (RP) scores are calculated for given genes to measure the regulation strength of TRs.The implemented model is:

wherekrepresents the count of peaks around the gene;dis the distance from the center of peakito the transcriptional start site (TSS) of the gene;is defined in the original study[20];mis the overall enrichment of the signal for the peak regions from the Encyclopedia of DNA Elements(ENCODE) database.
Step 3:selection of the TRs most relevant to drug response
In step 3.1,TR independence is estimated through Elastic-Net (EN) model.Many TRs have overlapping regulation effects with other TRs.To select the key regulators among the overlapping TRs,an EN method is conducted by regressing the RP scores on the partial correlations of all the genes.By using this step,RePhinetakes both effects of TFs and CRs into account simultaneously.The EN estimator [21] is defined as:

If multiple replicates or samples in different cell lines are available for the same TRt,RePhineonly chooses the replicate or sample with the largest statistical effect of association between the targets and the given drug (the most significant uniP) to represent the TRt(analogously to the approach of the RABIT method[17]).
To determine theαin the EN model,we tried different values ofαand found highly consistent results of TR selection (see File S1 and Table S2 for more details).Therefore,αis fixed to 0.8 to shorten the time of calculations.λis obtained from leave-one-out cross-validation where the parameter“lambda.1se”is used to avoid the overfitting.The R“glmnet” package is used in the calculation.
In step 3.2,a likelihood ratio test is utilized to assess the significance of estimated coefficients for both the EN model step(step 3.1)and the AL model step(step 1.2.2);uniPand multiPare calculated by comparing the goodness of fit of these two models with inclusion/exclusion of the selected variable.In the univariate setting,model 1:is compared to model 2:y=intercept.In multivariate setting,all predictors selected from AL or EN are regarded as“S”;thus,model 1:y=βS+interceptis compared to model 2:y=βS-x+intercept,wherexis the variable of interest.Variablesyandxin the AL and EN models are the same as those in the step 1.2.2 and step 3.1,respectively.The R “lrtest”package is used in this step.
In these procedures,uniPis used to measure the overall significance of associations between RP scores and partial correlation coefficients;multiPis applied to assess the independence of the associations.
Step 4:visualization of the association
In this step,an enrichment algorithm is additionally implemented to visualize the association and the enrichment patterns.PermutationPvalue (permuP) is used to assess significance of the enrichment.The details of the algorithm are as follows:
1) Rank theNgenes according to the partial correlation coefficients.
2)Normalize the vectorPreg(the RP scores)to a range[0,1]across the genes within the sample by dividing by the maximum values.
3) Evaluate values for the genes as belonging(hit)and not belonging (miss) to the TR targets weighted by the partial correlationrpartial.For the topigenes ranked by partial correlations:

whereNrepresents the gene count.
4) Estimate the maximum deviation ofDhit-Dmissfrom zero.For random distributions of TR targets,the enrichment score ES(TR)will be a relatively small value.In contrast,if targets with higher RP scores assemble at the top or bottom of the partial correlation list,the ES(TR)will be high.WhenPregis binary(0 or 1),ES(TR)reduces to the original gene set enrichment analysis(GSEA)ES(see File S1 for details).WhenPregis equal to 0 or 1 andkis simultaneously equal to 0,the ES(TR) will be the standard Kolmogorov-Smirnov test statistics [22].
5)Permute the gene labels and re-compute the ESs.Repeat 1000 times to obtain the distribution of ESrandom.The assessment ofPvalue is based on the positive or negative segments of the distribution of ESrandomdepending on the sign of ES(TR).
Pharmacologically-relevant patterns such as positiveassociation,negative-association,and non-association could be distinguished and visualized through the aforementioned formula(Figure S3).
Drug combination screening
The interaction between PLX4720 and GSK126 is measured by the combination index (CI) derived from CalcuSyn[23].CI=1 indicates additive effects;CI >1 and CI <1 indicate antagonism and synergism,respectively.
Results
RePhine has an improved performance in simulation data with added noises and CFs
To systematically examine whetherRePhinecan accurately identify the associations between the TRs and the drug response,its performance was evaluated in the simulation datasets.We comparedRePhinewith three commonly used methods:Pearson correlation (PC) analysis,logistic regression model,and GSEA (Figure S4).With the increase of CF counts and expression noise levels,RePhineshowed a significantly improved performance (Figures S4A-C).AlthoughRePhinedidn’t overperformed GSEA with the expression noise level increasing because both methods consider target information,RePhinehad an improved performance when noise was added to the RP scores of target information (Figures S4D-G).In addition,we also compared these methods in the scenario where multiple noises and CFs were added simultaneously to a balanced dataset and an imbalanced dataset,respectively(Figure S4H and I).RePhineexhibited higher area under the receiver operating characteristic (AUROC) values than the three commonly used methods in the balanced dataset when noise level and CF count increasing.Similarly,RePhineshowed higher area under the precision recall curve(AUPRC)values in the imbalanced dataset when noise level and CF count increasing.These simulation results clearly demonstrate the advantage ofRePhineover these commonly used methods(for more details of the comparison,see File S1).RePhineemploys a novel strategy with careful consideration of noises and CFs and enables robust identification of TRs.
RePhine enables identification of TRs with significant and non-significant correlation between their mRNA levels and drug response
When applied to pharmacogenomic data,RePhinecan identify TRs which have significant correlation between their mRNA expression levels and drug response.Furthermore,RePhinecan also identify candidate TRs which do not have significant correlation between their mRNA expression levels and drug response (Figure 2A and B).We appliedRePhineto data from CCLE and ENCODE and performed bothRePhineand PC analysis to compare the two methods which are based on two different biological assumptions.For each TR,two metrics were calculated:1)RePhinesignificant score(uniP)in log scale with direction(resistance or sensitivity) and 2) Pearson correlation coefficient (PCC) between its mRNA level and drug response.For the 24 drugs available in CCLE,the two metrics were positively correlated in most drugs (Figure 2A,Figure S5A),supporting the biological assumption that TRs influencing the target regulation by their mRNA activation or deficiency could also be effectively identified by targetbased methods such asRePhine.In the specific examples of erlotinib (a well-established anti-cancer targeted therapy drug agent [15]) and paclitaxel (a chemotherapy drug),RePhinesignificant scores (uniP) were highly correlated with the PCCs across all the TRs (Figure S5B and C).
How doesRePhineperform on TRs which do not have significant PC between their mRNA expression levels and drug response?We then chose erlotinib to assess this issue.First,we divided the TRs into 3 groups.Group 1 contained the PC-correlated-only TRs (Figure 2B,green set;having significant PC between their mRNA expression levels and drug response but notRePhinesignificant uniP).Group 2 contained theRePhine-correlated-only TRs(Figure 2B,blue and purple sets;havingRePhinesignificant uniPbetween their mRNA expression levels and drug response but not significant PC);TRs in the purple set additionally hadRePhinesignificant multiP(within theRePhinemultivariate EN cutoff multiP<0.005),and thus were predicted to be involved in the drug response independently (namedRePhine-correlated-only independent TRs) (Figure 2B,Figure S5D).Group 3 contained the PC-RePhine-sharedcorrelated TRs(Figure 2B,red set,common TR candidates).
We then performed protein-protein interaction (PPI)analysis to investigate the biological connections among the TR candidates within each set.RePhine-correlated-only independent TRs (purple set) had more significantly enriched PPIs than PC-correlated-only TRs (Table S3).Both theRePhine-correlated-only independent TRs (purple set)and the PC-RePhine-shared-correlated TRs (red set) had over-represented pairwise PPIs (Figure 2C,Figure S6A).The pairwise PPIs achieved higher enrichment by pooling such TRs together (P=2.58E-10,derived from STRING database;Figure 2C and D;Table S3),suggesting tighter biological connections.In contrast,there were less significantly enriched PPIs observed among the CAcorrelation-only TRs (green set) and among the CAcorrelation-only and PC-RePhine-shared-correlated TRs(green and red sets) (Figure S6B and C;Table S3),suggesting unrelated and random connections between them.In addition,FOS and STAT3,two known biomarkers of erlotinib [16,24-26],had tight interactions with the other candidates (Figure 2C and D).
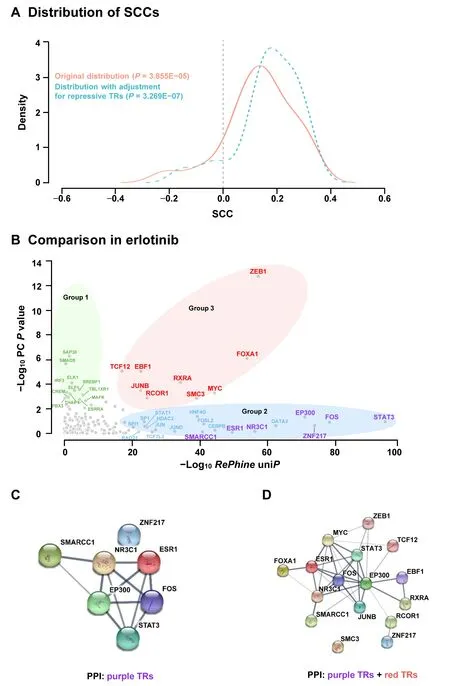
Figure 2 Comparison of the results between RePhine and PC analysis in CCLE data
RePhine has an improved performance in CCLE dataset
To comprehensively evaluate theRePhineperformance in real data,we next conducted PPI enrichment analyses to the TR candidates from PC analysis,GSEA,andRePhinefor all drugs in CCLE dataset (Table S4).
RePhine-correlated independent TRs (having both significant uniPand significant multiP;see Method for details)displayed a significantly higher PPI enrichment than PCcorrelated TRs (P=0.003575,derived from STRING database;Table S4).However,this enrichment was not significantly higher than that of GSEA-correlated TRs.The results are reasonable because GSEA candidates contain redundant TRs such as subunits of the same complex,which tend to have more PPIs butRePhineonly identifies the independent TRs among these redundant TRs.To test this notion,we compared theRePhine-correlated TRs that only had significant uniP(where functionally redundant TRs were not removed)with GSEA-correlated TRs.The results showed that theRePhine-correlated TRs had a significantly higher PPI enrichment than GSEA-correlated TRs (P=6.52E-05,derived from STRING database;Table S4).The significantly higher PPI enrichment ofRePhine-correlated TRs suggests that TRs identified byRePhinehave tighter biological connections and functional consistency.
To further evaluate the effectiveness ofRePhine,we next performed PPI enrichment analyses (Table S4) and literature searching for CCLE drugs with marketing approval(Table S5) to compareRePhinewith a published effective method “DoRothEA” [27].Both methods were applied to CCLE data.RePhine-correlated independent TRs had a significantly higher PPI enrichment (P=0.01488,pairedt-test;Table S4),and the count of publication-consistent TRs(the true positives)ofRePhinewas higher than that of DoRothEA (38 hitsvs.12 hits;P=1.078E-04,Fisher’s test).In contrast,the counts of the false negatives and the candidates without literature support ofRePhinewere lower(16 hitsvs.36 hits,P=0.004141,Fisher’s test;55 hitsvs.80 hits,P=0.00942,Fisher’s test;Table S5).The counts of the contrary predictions of both methods (where the predicted resistance or sensitivity were contrary to the publications)were similar(7 hitsvs.13 hits).
RePhine effectively clusters drugs with similar action mechanisms
Next,we took advantage ofRePhineto explore the relationship between TR regulation and drug response.We defined the TR significance profiles ofRePhinecorrelation(-Log10uniPwith direction)across all CCLE drugs as TR response signatures.The response signatures reflect whether each drug has a specific TR response pattern.We computed the response signatures for all ENCODE TRs(n=160).Notably,we did not filter TRs in this procedure.RePhineuniPvalues were calculated for all 160 TRs.
Our results suggest that drugs with similar action mechanisms tend to have similar TR signatures andRePhinecan effectively cluster drugs with similar action mechanisms together.RePhine-based clustering separated the drugs into three clusters (Figure 3A).In cluster 1,all chemotherapy drugs as well as an HDAC inhibitor (panobinostat) were clustered together but away from other targeted therapy drugs.In cluster 3,drugs targeting EGFR(erlotinib,AZD0530,lapatinib,and ZD-6474)[28-30]were clustered together with an HSP90 inhibitor (17-AAG).In cluster 2,there were two subgroups.Cluster 2.1 contained two ALK inhibitors (PF-2341066 and TAE684),an ABL inhibitor (nilotinib),a CD4/6 inhibitor (PD-0332991),an IGF-1R inhibitor (AEW541),and two multi-kinase inhibitors (sorafenib and TKI258) [28];in cluster 2.2,two MEK inhibitors (PD-0325901 and AZD6244),two RAF inhibitors (PLX4720 and RAF265),an MET inhibitor(PHA-665752),which were related to MAPK signaling[31],were clustered.All the drugs in cluster 2 were targeted therapy drugs[28].In addition,clustering based onRePhinesignatures had an improved performance compared with GSEA-based clustering in the mechanism-focused separation of the CCLE drugs(Figure 3A,Figure S7;File S1).
Hierarchical clustering analysis further separated 160 TRs into five clusters,which were associated with different types of therapies(Table 1).Each cluster showed enriched pairwise PPIs and functional consistency(Figure 3B;Table S6).For example,TR response signatures in chemotherapy drugs were associated with cell cycle as expected (Figure 3A and B),indicating that the cell cycle-related TRs could significantly regulate chemotherapy response[32];the TRs in cluster 1 enriched in AP1 pathway were positively correlated with the response to both BRAF inhibitor and EGFR inhibitors.The TRs in cluster 5 that are related to EGF response and IFN-related pathways were only associated with the response to EGFR inhibitors (Figure 3A and B).
How can this TR clustering inform regarding response to anti-PD-1 immunotherapy? To answer this question,we integratedin vivoanti-PD-1 CRISPR screening data and patient-level anti-PD-1 response data from two previous studies [33,34].By comparing these TR clusters with CRISPR screening candidates,we found that the top 5 TRs(STAT1,SMARCA4,STAT2,GTF2F1,SMARCC1)whose loss-of-function would trigger the resistance to anti-PD-1 therapy were enriched in cluster 5 (P=0.034,one-tail Fisher’s test;Figure 3C).To validate this observation,we explored the associations of these TRs with anti-PD-1 drug response in the patient cohorts [33].Although these TRs were not observed to be differentially expressed between responders and non-responders,mutations of the genes encoding TRs in cluster 5 exclusively resulted in lower probability of complete response to anti-PD-1 therapy than that of partial response and progressive disease,which was identified through a multivariate logistical analysis by accounting for mutation loading(P=0.0144,Table S7).
These results suggest that TRs particularly associated with response to EGFR inhibitors (Table 1,cluster 5) are also linked with anti-PD-1 effect [34].Interestingly,it has been reported that EGFR pathways can positively regulate the activation of PD-1/PD-L1 pathway[35,36];such studies may explain why there is a common TR response signature between EGFR inhibitors and anti-PD-1.
Identification of EZH2 as a BRAF inhibitor resistancerelated TR
PLX4720,the precursor of vemurafenib (PLX4032),is an ATP-competitive BRAF inhibitor.Resistance to BRAF inhibitors may rapidly develop in patients,but the mechanisms underlying this resistance are not fully understood [37-39].We appliedRePhineto PLX4720 using CCLE data and ENCODE ChIP-seq data,and identified GTF3C2,YY1,ESR1,E2F1,MYC,GATA3,RBBP5,EZH2,E2F4,ZEB1,and ZNF217 as theRePhinenegatively-correlated independent TRs (RePhinecoefficient <0,uniP<1E-5,multiP<0.005,Figure 4A;Table S8).Among them,EZH2,E2F4,ZEB1,and ZNF217 are primarily transcription repressors (Table S9).On the other hand,activations of SPI1,CEBPB,and EP300 were determined as PLX4720 sensitivity predictors (RePhinecoefficient >0).

Table 1 Hierarchical clustering of TRs associated with different types of therapies
To validate all these candidates obtained fromRePhine,we integrated the CRISPR-meditated gene knockout screening results for these candidates in A375 cells with PLX4032 treatment[37].We used the MAGeCK method to interpret the CRISPR results [40].Beta scores from MAGeCK indicate sgRNA abundances in the screen and the differences of beta scores between treatment and control reflect the effects of the drug on cell survival after gene knockout [40].RePhine-identified candidates including resistant-related TRs as well as sensitive-related TRs were all observed to correspond well with the differences of beta scores from CRISPR results (Figure 4A).These results indicate thatRePhinecan effectively identify drug responserelated TRs.
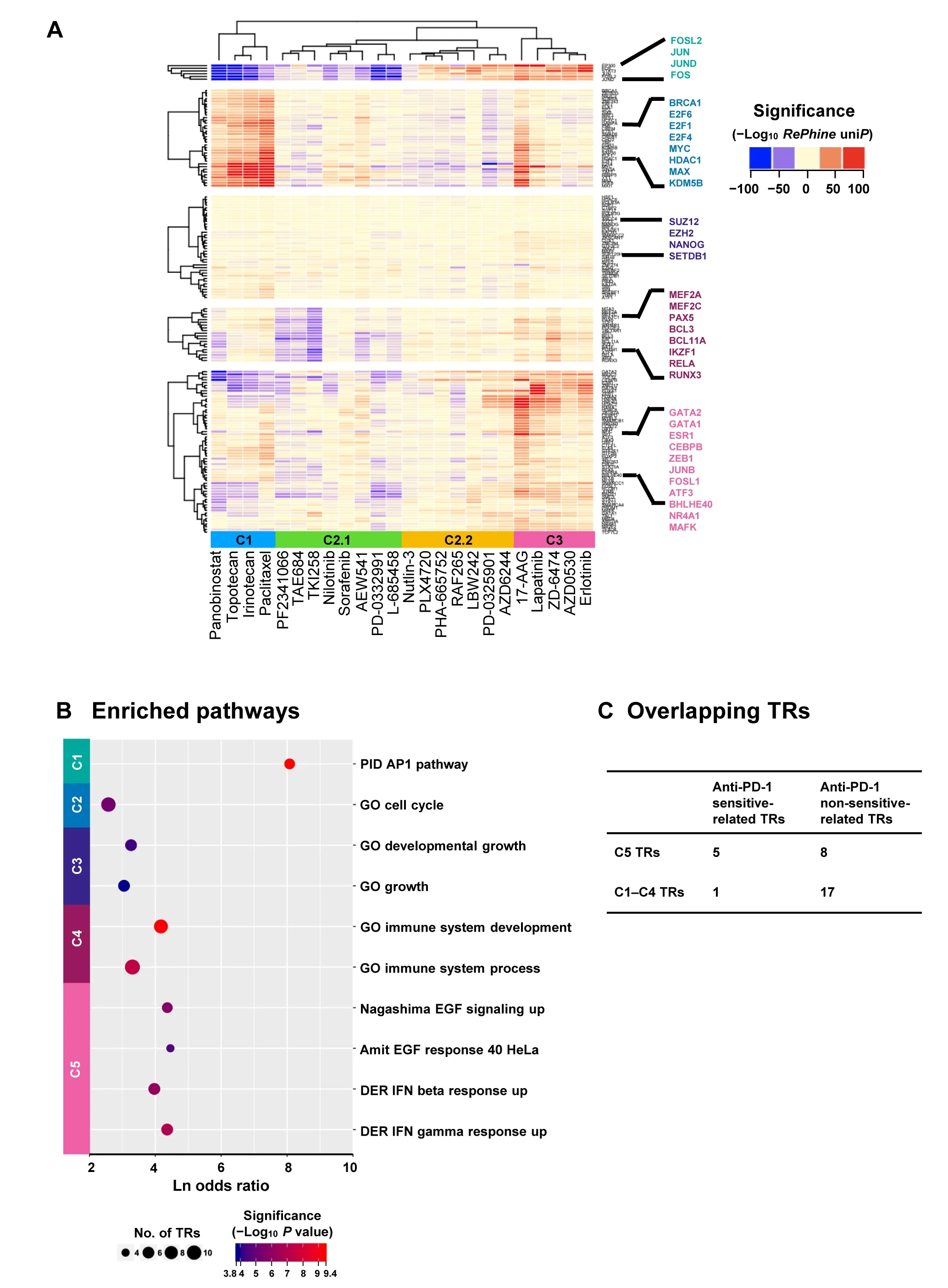
Figure 3 RePhine-characterized TR response signatures of drug clusters and functional enrichment
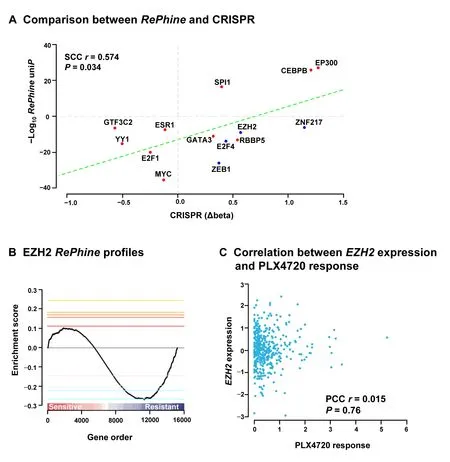
Figure 4 EZH2 is predicted as a BRAF inhibitor resistance-related TR by RePhine
Validation of EZH2 as a PLX4720 resistance-related TR by drug experiments
OurRePhineresults showed that EZH2 is related to PLX4720 resistance.We observed that EZH2 had negativeRePhinecorrelation with the PLX4720 response (Figure 4B);however,its mRNA levels did not exhibit any correlation with the PLX4720 response (Figure 4C).Therefore,we performed experimental validation of thisRePhineprediction.
To experimentally validate theRePhineprediction of EZH2,we performed drug combination experiments with GSK126 and PLX4720.GSK126 is a potent and highly selective EZH2 inhibitor[41].In bothBRAFV600E mutant cell lines,A375 and SK-HEP-1,treatment with PLX4720 and GSK126 simultaneously resulted in antagonistic inhibitory effects at most dosage combinations(average CI=3.281 for A375;average CI=1.833;Figure 5A-D).In contrast,in theBRAFwild-type cell line JHH-7,there was no strong antagonistic interaction at most dosage combinations (average CI=1.021),which suggests that the antagonistic effect is BRAF activation-dependent (Figure S8A and B).Contrary to our expectations,PLX4720 and GSK126 showed slight synergistic effect at high GSK126 dosages in SK-HEP-1 and JHH-7(control)cell lines.This is possibly due to EZH2 being functional at the high dosage or existence of off-target pathway inhibition [42].Similar antagonistic interactions were observed in two previous studies:experiments in cell lines [43] andin vivomouse models [44].It was worth pointing out that such studies claimed synergistic effect between BRAF inhibitors and EZH2 inhibitors in a subset of cancers that hadEZH2amplification or gain-of-function mutations which led to redistribution of H3K27me3 [44].Their control experiments(in cell lines with wild-typeEZH2) supported our finding,but the studies’ authors did not comment on their control findings.
Validation of EZH2 as a PLX4720 resistance-related TR by bioinformatics analyses
We next validated the EZH2 role in PLX4720 resistance by analyzing the EZH2 ChIP-seq targets selected byRePhine,the differentially expressed genes in PLX4720-resistant cell lines,the relationship between PLX4720 response and PRC2 gene mutations in the CCLE data,and the effect of PRC2 deficiency on clinical outcomes in patients.
Given that EZH2 is a methyltransferase for H3K27[42],we analyzed the EZH2 ChIP-seq targets selected byRePhine(Table S10).Besides the H3K27me3 signatures,the EZH2 ChIP-seq targets were enriched in several cancerrelated pathways (Figure S8C;Table S10).Interestingly,knockout of genes encoding PRC2 essential subunits and other H3K27me3-related TRs,including ZNF217 [45,46],also drove the cells to become comparatively resistant to BRAF inhibitor treatment according to the CRISPRmediated gene knockout data (Figure S8D;Table S11).
We also discovered thatEZH2and genes encoding other PRC2 subunits were significantly down-regulated in the BRAF inhibitor-resistant cells in two independent expression datasets GSE68840 and GSE68599(Figure 5E).
In the analysis of PLX4720 response and PRC2 gene mutations in the CCLE data,we found that cell lines containing mutations in bothBRAFand H3K27me3-related genes had significantly lower PLX4720 response than those with onlyBRAFmutations (P=0.044,t-test;Figure S8E).
In TCGA skin cutaneous melanoma (SKCM) patient cohort dataset,where around half of the patients gainedBRAFmutations,we investigated whether PRC2 deficiency affects clinical outcomes in patients.We defined an activity score for each patient to evaluate the activity of PRC2.Patients with relatively lower PRC2 activity scores had worse outcomes than those having higher scores in theBRAFmutant group (hazard ratio=0.62,P=0.041,95%confidence interval:0.40-0.98;Figure 5F).In contrast,the PRC2 activity scores were not predictive of overall survivals in theBRAFwild-type group(hazard ratio=1.14,P=0.516,95% confidence interval:0.76-1.71;Figure 5G),consistent with the results of the univariate Cox regression analysis(Figure S8F).

Figure 5 Validation of EZH2 as a BRAF inhibitor resistance-related TR
Discussion
Due to low mRNA abundance of TRs and complexity of biological regulation mechanisms,detecting the linkage between TRs and drug response is still challenging.In this study,RePhinewas developed to effectively perform three main tasks:1)an integrative analysis on ChIP-seq targets to produce TR identification robust toward noise and complicated protein modifications,2)an accurate measurement of correlation patterns by adjusting all potential CFs that are not under the impact of TR regulation,and 3) application of quantitative and informative target inference by considering both ChIP-seq signals and the distances from peaks to the targets to achieve a better evaluation of the associations.
There are still areas for further improvement.First,although significant correlations between TRs and pharmacological profiles could be detected throughRePhineby exploring targets’ profiles,the relationship may not be causal.Some upstream regulators or kinases may exist that influence the drug response and simultaneously regulate downstream TRs.Therefore,TR target analyses and TR knockout or activation experiments combined with drug response examination are also required to validate the causal relationship.Second,becauseRePhineidentifies drug response-related TRs through the targets,it is restricted to the cases where reliable target information is available.Lack of ChIP-seq data or ChIP-seq data with too few targets would mislead the identification of TRs.Hence,some existing target prediction algorithms could be exploited and complemented to facilitate target inference.Third,detection of acquired resistance is limited due to lack of post-treatment data in CCLE [28].It is not trivial to detect the secondary alterations in response to drug treatment,which may elucidate why secondary resistance to erlotinib through acquired STAT3 activation[47]could not be detected byRePhine.In addition,due to lack of posttreatment data,it is hard to integrate the effect of drug perturbations on genes[28,48].However,the pre-treatment correlation identified byRePhinemay still be relevant to such drug influence on TRs.For example,FOS is selectively up-regulated by EGF stimulation and inhibited by EGFR TKI treatment in sensitive cells rather than in resistant cells [16].If FOS could not be inhibited by EGFR TKI,the cells with higher FOS levels would not be sensitive to EGFR inhibition,and there would be no correlation between FOS activity and drug response.Nevertheless,such associations need further evaluation when post-treatment data are available.
RePhinehas been further applied to an independent unpublished liver cancer dataset containing RNA-seq data,copy number information,and DNA sequencing data from more than 50 primary liver cancer cells coupled with pharmacological profiles for nearly 100 anti-cancer drugs [49].Our novel identification,which has been validated by experiments,is that MYC promotion could independently and significantly increase the response of three drugs:BI-2536,PF-562271,and panobinostat.However,MYCmRNA did not show any correlations with the pharmacological profiles (Figure S9).The positive results obtained by applyingRePhineto this liver cancer dataset further suggest thatRePhineis an effective method for identifying drug response-related TRs and could be used in other independent pharmacogenomic data.
Code availability
RePhine,which is implemented as an R package and accompanied by a user guide,is available at https://github.com/coexps/RePhine.RP scores,TCGA differentially expressed gene sets,modified Python scripts of RP score calculation,and R code for simulation and application for CCLE data are also available at https://github.com/coexps/RePhine.
CRediT author statement
Xujun Wang:Conceptualization,Methodology,Formal analysis,Software,Writing-original draft,Writing-review&editing.Zhengtao Zhang:Validation,Investigation.Wenyi Qin:Writing -original draft,Writing -review &editing.Shiyi Liu:Software.Cong Liu:Methodology.Georgi Z.Genchev:Conceptualization,Methodology,Formal analysis,Writing-original draft,Writing-review&editing.Lijian Hui:Validation,Investigation.Hongyu Zhao:Conceptualization,Methodology,Writing -original draft,Writing -review &editing,Supervision.Hui Lu:Conceptualization,Methodology,Formal analysis,Writingoriginal draft,Writing -review &editing,Supervision.All authors have read and approved the manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
We thank members in Department of Biostatistics,Yale School of Public Health and SJTU-Yale Joint Center for Biostatistics for comments and discussion.We also thank the members in Lijian Hui group for experimental guidance and comments.This work is supported by the National Key R&D Program of China (2018YFC0910500),the Neil Shen’s SJTU Medical Research Fund,the SJTU-Yale Collaborative Research Seed Fund,the National Natural Science Foundation of China (Grant Nos.31370751 and 31728012),the Shanghai Municipal Commission of Health and Family Planning(Grant No.20144Y0179),the Science and Technology Commission of Shanghai Municipality(STCSM) (Grant No.17DZ 22512000),the Shanghai Municipal Science and Technology Major Project (Grant No.2018SHZDZX01),and the Key Laboratory of Computational Neuroscience and Brain-Inspired Intelligence(LCNBI) and ZJLab.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.09.008.
ORCID
0000-0002-7152-611X(Xujun Wang)
0000-0003-0746-8004 (Zhengtao Zhang)
0000-0001-5460-0325 (Wenyi Qin)
0000-0002-2157-8667 (Shiyi Liu)
0000-0001-6024-3037 (Cong Liu)
0000-0002-3281-9104 (Georgi Z.Genchev)
0000-0002-6213-6187 (Lijian Hui)
0000-0003-1195-9607(Hongyu Zhao)
0000-0001-8347-0830 (Hui Lu)
 Genomics,Proteomics & Bioinformatics2021年4期
Genomics,Proteomics & Bioinformatics2021年4期
- Genomics,Proteomics & Bioinformatics的其它文章
- SPA:A Quantitation Strategy for MS Data in Patient-derived Xenograft Models
- NOGEA:A Network-oriented Gene Entropy Approach for Dissecting Disease Comorbidity and Drug Repositioning
- DeepCAPE:A Deep Convolutional Neural Network for the Accurate Prediction of Enhancers
- The Genome Sequence Archive Family:Toward Explosive Data Growth and Diverse Data Types
- Genome Warehouse:A Public Repository Housing Genomescale Data
- REVA as A Well-curated Database for Human Expressionmodulating Variants