
Research and Reference of the Authorized Generic Drug System in the US
2021-03-22 07:50:58WangHongweiYangYue
亚洲社会药学杂志 2021年1期
Wang Hongwei ,Yang Yue
(1.School of Business Administration,Shenyang Pharmaceutical University,Shenyang 110016,China;2.International Center for Food and Drug Policy and Law Research,Shenyang Pharmaceutical University,Shenyang 110016,China)
Abstract Objective To study the significance of authorized generic drugs due to the successive relevant documents issued by China’s government in recent years,which clearly stipulated that China should establish a drug patent linkage system.Methods The authorized generic drugs play an important role in keeping the balance between brandname drugs and generic drugs in the US.Therefore,this system was investigated,focusing on its difference from independent generics,marketing procedures,application in patent litigation and legitimacy analysis through case evidence.This analogy analysis could provide a reference for the research of authorized generic drug systems in China.Results and Conclusion As an important factor affecting the balance between brand-name drugs and generic drugs,authorized generic drugs should be comprehensively analyzed and discussed,and a suitable system for China’s national conditions should be established by referring to the experiences of the US.
Keywords:authorized generic drug; patent linkage system; drug; settlement; patent litigation
In recent years,China’s drug review and approval system have continuously deepened its reform,and policies of drug and medical device innovation have been introduced in succession.Among them,the patent linkage system which balances the interests of brand-name drugs and generic pharmaceutical enterprises has been widely concerned.The General Office of the Communist Party of China and National Medical Products Administration issued relevant documents,clearly stipulating that China should establish a drug patent linkage system.It is a system that links the drug registration of the drug regulatory department with the work of the judicial department to resolve patent disputes.Since the authorized generic drug system and relevant regulations will have a significant impact on generic drug companies to file patent declaration,drug patent litigation and the exclusive period of generic drugs,we should focus on the advantages and disadvantages of the system before establishing it in China.In the US,authorized generic drug is widely used as a tool for agreements between brand-name companies and generic drugs companies,which plays an important role in the health care system.This paper focuses on the study of the authorized generic drug system in the US in order to provide a reference for the establishment of this system in China.
1 Comparison of brand-name drugs,authorized generic drugs and independent generic drugs
Brand-name drugs,also known as innovative drugs,refer to drugs with independent intellectual property rights,which can only be produced and sold by companies with such patents.Compared with generic drugs,brand-name drugs emphasize novel chemical structures or new therapeutic uses,which have not been reported in previous research literature or patents.Brand-name drugs provide patients with new solutions for the treatment of diseases without satisfactory methods in the market.Its development is a time consuming and expensive process.In order to obtain regulatory approval,brand-name drugs must show obvious advantages over existing drugs.In China,brand-name drugs refer to Class I new drugs,that is, drugs that have not been marketed at home and abroad.
According to different production standards,generic drugs are divided into independent generic drugs (IGs) and authorized generic drugs (AGs)[1].It means they belong to different concepts and have no trade names.21 CFR Section 314.3 defines AG as follows:Authorized generic drug is a listed drug,as defined in this section,that has been approved under section 505 (c) of the “Federal Food,Drug,and Cosmetic Act” and is marketed,sold,or distributed directly or indirectly to the retail class of trade with labeling,packaging (other than repackaging as the listed drug in blister packs,unit doses,or similar packaging for use in institutions),product code,labeler code,trade name,or trademark that differs from that of the listed drug[2].
It can be seen from the above definition that AG is a kind of drug that is marketed under the new drug application (NDA) of an approved brand-name drug and is essentially identical to the brand-name drug,except that the trade name is not used on the label.Unlike an IG distribution channel,AG can be sold through a subsidiary of brand-name drug manufacturer,enter into a contract with a distributor to market AG,or authorize a generic drug manufacturer to sell AG.A listed AG does not need to submit a separate NDA.FDA requires that if the NDA holders listed AG,they shall notify FDA via an annual report.NDA holders can market AG and brand-name drugs at the same time.AG can be listed at any time before and after the expiration of the patent,but it will usually be listed during the exclusive period as a strategy for the brandname drug manufacturer to research a settlement agreement with the generic manufacturer.Since AG applied for marketing under the approved drug’s NDA and did not submit a separate abbreviated new drug application (ANDA),it was not included in the Orange Book by FDA.Instead,a separate list database on AG was established and updated quarterly.The differences between AG and IG are shown in Table 1.

Table 1 Comparison between AGs and IGs
2 Study on regulatory path and marketing authorization process of AG
2.1 Regulatory path of AG
AG has two potential regulatory paths.When AG complies with the NDA of the brand-name drug (the formula,manufacturing process and manufacturing site,etc.have not changed),the only change is the new label of generic drugs,and the NDA holder can inform FDA of marketing by submitting an annual report.According to 21 CFR 314.70,when the label change of AG does not involve the change of the identification content,there is a potential minor impact on the safety and effectiveness of drugs,which is a minor change and can be sold on the market without approval[3].
The second regulatory approach involves changes in manufacturing sites.When manufacturers produce AGs with the same formula,manufacturing process and testing,etc.and NDA holders add new manufacturing sites to NDA,it involves the supervision of the manufacturing site.The change can be applied through prior approval supplement (PAS)or a supplemental application 30 days before the product distribution (CBE-30 supplement).According to Article 351 (a) of the “Federal Food,Drug and Cosmetic Act” FD&CA,the cGMP status of the new establishment shall be taken into account in the change of the manufacturing site:if it is transferred to a site that has never been inspected by FDA or the cGMP inspection is unqualified,PAS report must be submitted; if the cGMP inspection is qualified,the new site or production termination time is within 2 years when the site is changed,only the CBE-30 report is required,otherwise the PAS report must be submitted[4].
In summary,when the manufacturing site changes,the brand-name drug manufacturer must submit the corresponding change report and wait for 30 days or more before marketing AG.Therefore,the brand-name drug manufacturers usually change product labels when marketing AG,so as to seize market share at any time.
2.2 Procedures of marketing notification through the annual report
Since authorized generic drugs are essentially the same as brand-name drugs with patent rights,and their production licenses are the same as the brand-name drugs that have been approved by the drug regulatory department,the marketing or sales of generic drugs no longer need to undergo substantial review by the drug administration department.It only needs to submit the application materials of the brand-name drugs to FDA for filing,so as to obtain the marketing approval of the authorized generic drug.The filing method is to inform FDA through an annual report.
In the US,each NDA and ANDA applicant must submit an annual report,which will include all information about the approved drugs,such as:distribution information,labeling,changes in chemistry,manufacturing and control,clinical data,non-clinical laboratory data,and any other information about AG.The information and method of submitting AG specified in the annual report in 21 CFR section 314.81(b) (2) are shown as follows[5].
If applicable,the date that each authorized generic drug entered the market,stopped being distributed,and the corresponding trade name should be submitted.Each authorized generic drug with a different dosage form and/or strength should be listed separately.The first annual report submitted on or after January 25,2010 must include the information about AG that was marketed during the time period covered by an annual report submitted after January 1,1999.If information is included in the annual report with respect to any authorized generic drug,a copy of that part of the annual report must be sent to the Office of New Drug Quality Assessment (ONDQA),Center for Drug Evaluation and Research of FDA.However,when FDA requires the annual report to be submitted in electronic format,the information about AG must be submitted in the specified electronic format as part of the annual report,rather than separately.Unlike brandname drugs and IGs,which need to go through a complex substantive review to obtain the marketing authorization,AGs can not only save the cost of submitting generic application,but also mean that once the brand-name drug manufacturer determines that the authorized generic drugs can make profits,AGs can be marketed at any time (i.e.not limited by the brand-name drug patents).This is a faster way to market than independent generics[1].The comparison of approval of brand-name drugs,generic drugs and AGs is shown in Table 2.

Table 2 Comparison of approval of brand-name drugs,generic drugs and AGs
In the US,FDA approval is required for the voluntary withdrawal of approved drugs from the market[6].However,in the practice of AGs,except that the contents specified in the annual report should be reported to FDA,the decision to stop marketing AGs does not require FDA approval.
2.3 Listing database of AGs in FDA
FDA requires NDA holders to clearly report that they have listed AGs in descriptive language when submitting their annual report.The FDA has published a list of all AGs,namely,“FDA Listing of Authorized Generics”[7].The column “Authorized generic drug market time” reflects the date of the first report of AGs in the annual report,but due to the large amount of data,the accuracy of the information in the list cannot be guaranteed.Besides,because there is no mandatory requirement for the applicant to submit a decision to withdraw the listing,FDA cannot know the date when it suspends sales.As a result,the information about AGs available from the list is limited and incomplete.Compared with the current reporting FDA system,the establishment of a process specific to AGs approval will help to build a more comprehensive database[8].
3 AGs and patent litigation settlement
According to “Drug Price Competition and Patent Term Restoration Act” (also known as “Hatch-Waxman Act”),when the first filer challenges a patent for a brand-name drug’s patent,FDA may not approve additional generic drugs for another 180 days after the generic product is marketed (according to the section 355(j) (5) (b) (iv) of 21 U.S.C.,except that two or more applicants submit their applications on the same day for a “shared exclusivity period”).The US Congress created this exclusivity period to encourage generic drugs to be launched as soon as possible by challenging invalid or non-infringing patents.After the first generic drug is sold on the market,brandname drug manufacturers will quickly face a loss of revenue during the exclusive period.Normally,they will increase their market share by listing AGs.Since AGs are marketed based on NDAs that have been approved by FDA for brand-name drugs,they can be marketed during the exclusive period of the first generic drug.During the 180-day exclusivity period,competition from AGs is likely to reduce generic prices and revenue for generic manufacturers,so AGs have a significant impact in the first six months of generic competition.
AGs can be used to resolve patent infringement lawsuits between brand-name drugs and independent generic drug manufacturers.The judicial determination of patent invalidation can have a serious impact on the revenue of brand-name drug manufacturers,while patent litigation is also an uncertain risk for generic drug manufacturers.By solving patent litigation through AG strategy,brand-name drug manufacturers can better manage risks and provide more fund to support their research and development activities.Generic manufacturers can also avoid spending money in uncertain litigation or seeking FDA approval for their ANDA.At the same time,if generic manufacturers are authorized to distribute AGs,they can expand their product lines to gain production experience and a first-mover advantage in the generic drug market[9].
3.1 Application of AGs in patent dispute
Brand-name drug manufacturers and generic drug manufacturers are traditionally fierce competitors.In recent years,pay-for-delay settlement has become a way for both sides to resolve the patent dispute.A pay-for-delay settlement is an agreement signed between brand-name drug manufacturers and generic drug manufacturers,which includes both the compensation to generic drug manufacturers and the restrictions on generic drug manufacturers from marketing their products[10].
The first generic applicant who successfully challenges the patent of paragraph IV (PIV)certification will be granted a 180-day exclusive period,during which the market duopoly will maintain a high price of the drugs,thus encouraging the generic manufacturers to bear the risk of legal proceedings to challenge illegal or weak patent.But brand-name manufacturers can respond on the PIV certification challenge by f illing patent lawsuits.A “30-month stay”period will start after the brand-name manufacturer f iles a lawsuit,creating an opportunity for both parties to use the pay-for-delay settlement[10].
Common patent settlements typically fall into the following categories:the f irst is reverse payment agreement,also known as the payment agreement,which means that the brand-name drug manufacturers pay corresponding fees to generic drug manufacturers who agree to produce or market their generic drug when the NDA holder’s patent expires[11].The second is the agreement between the brand-name drug manufacturers and generic drug manufacturers,which authorizes the generic drug applicant to use the patent of the NDA holder before the patent expires.As to another popular settlement,brand-name drug manufacturers would make “no AG” commitments as a way to compensate for generic drug manufacturers who agree to delay the launch of their products.In the 2011 report entitled “Authorized Generic Drugs:Short-term Effects and Long-term Impact”,US Federal Trade Commission noted that about 25% of the settlement reached between f iscal years 2004 and 2010 contained “no AG” provisions[12].This paper focuses on the types and applications of “no AG”settlements.
3.2 Types of settlements related to AG
Brand-name drug manufacturers and generic drug manufacturers can maximize their net present value of their revenue by entering into agreements that provide no competitive AG[13].The increase of brand-name drug manufacturers’ revenue is due to the delay of generic drug marketing when they promise not to market AG,while the increase of generic drug manufacturers’ revenue is due to the fact that their ANDA generic products will not face the competition from original drug manufactures’ AG.The specific terms of the “no AG” agreements are diff erent.There are a variety of compensations to reach settlement terms.Three main types are shown in Fig.1.
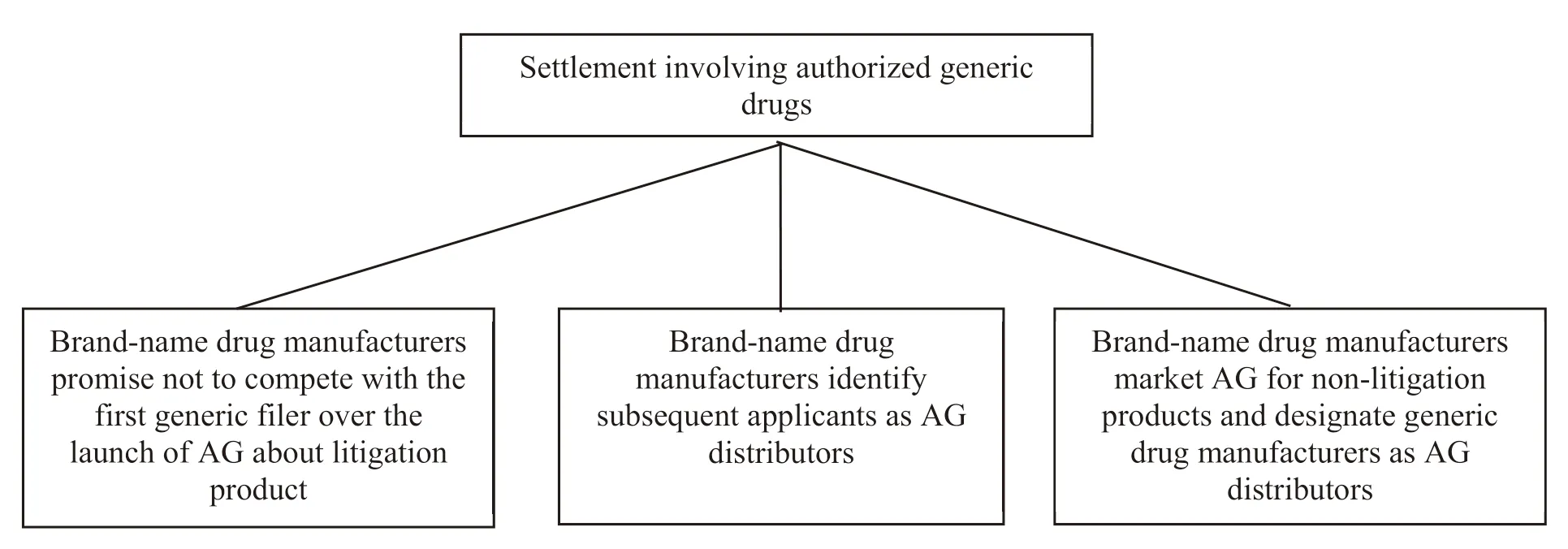
Fig.1 Types of settlements related to AG
The different compensations for the settlement are as follows.
(1) A clear commitment to unlisted AG.According to the final report of Federal Trade Commission (FTC) on authorized generic drugs,the period of “no AG” under the settlement that brandname manufacturers agree with ranges from 10 days to 45.5 months,which is related to the sales of the product.If the products have low sales,some settlements keep AG off the market for up to two years or more.If the products have larger sales,the limit on AG marketing competition is rarely longer than six months (a 180-day exclusivity period)[12].
(2) Brand-name drug manufacturers designate subsequent applicants as AG distributors of their products.To resolve patent lawsuit,brand-name drug manufacturers may seek to reach a settlement with a subsequent applicant.It means they try to market AG for litigation products during the exclusivity period of the first generic filer.However,brand-name drug manufacturers usually publicly announce the relevant terms of their settlements with subsequent applicants(usually trade secrets),and use the settlement as a tool to negotiate with the first generic applicant.There is a strong incentive for brand-name drug manufacturers to delay the exclusivity of applicants for ANDA with the PIV certification.
(3) Brand-name drug manufacturers designate generic drug manufacturer as AG distributor of another non-litigation product.The brand-name drug manufacturer and generic drug manufacturer may reach an agreement,which means the brandname drug manufacturer agrees not to market AG to compete with the first generic filer,but it can market the AG of non-litigation products and sign distribution contract with the generic drug applicant for sales.Such agreements can give generic drug manufacturers a percentage of revenue from the sales of other AG products.
This settlement can provide additional revenue to generic manufacturers and they agree to delay the launch of disputed products.For example,brand-name drug manufacturers agree not to compete against the first filer with AG,but also allow the generic drug manufacturer to distribute its other AG products.The agreement ensures that the generic drug applicant can have a 180-day exclusive period as the first generic drug filer for the disputed product.Meanwhile,the applicant can market another product as an AG distributor within a 180-day exclusive period.
3.3 Analysis of the impact of AG settlement on consumers
The “no AG” settlement is a way to solve the patent problems of brand-name drug manufactures and generic drug manufactures.Though it can benefit both parties accordingly,it is a kind of damage to the interests of consumers.The damage mainly includes two aspects:(1) Firstly,consumers are not able to obtain generic drugs with relatively low prices at an early time.Because the price of generic drugs is usually much lower than that of brand-name drugs.If there is no competition in the pharmaceutical market,the total cost of prescription drugs will greatly increase in a few months[14]. (2) Secondly,as the brand-name drug manufacturers agree not to market the AG in 180-day exclusive period and compete with the independent generic drugs,consumers cannot get the additional price discount brought by the AG competition in the exclusive period[15].
4 AG and the Medicaid drug rebate program
In 1990,the Congress of USA passed the“Omnibus Budget Reconciliation Act” of 1990,which established the Medicaid drug rebate program.That is,drug manufacturers participating in the Medicaid program are required to agree to provide a discount agreement to the Department of Health and Human Services (HHS) according to the terms of the standard national discount agreement.Meanwhile,part of the price discount of each drug provided to Medicaid will be returned to the state government in exchange for the federal Medicare reimbursements for the outpatient drug fees paid by the patients[16].
The “Omnibus Budget Reconciliation Act”established the discount schemes for brand-name drugs and generic drugs.A brand-name drug manufacturer is required to refund an amount equal to the product of the total number of units of each dosage form paid for under the Medicaid state plan times the greater of (i) minimum rate discount amount (currently 23.1% of average manufacturer price (AMP)) and(ii) the diff erence between AMP and best price (BP).The best price refers to the lowest price provided by the manufacturer to any wholesaler,retailer,supplier,health care organization,non-profit entity or government entity in the US during the discount period.The discount rate for generic drugs is 15.1% of AMP[17].
With the prevalence of AGs,some generic companies believe that there is a loophole in the Act,that is,the calculation of AG is not included in AMP and BP of brand-name drugs.Congress solved this problem by issuing the “Deficit Reduction Act”(DRA).Section 6 003 of the DRA stipulates that the def inition of the best price shall be modif ied to include the price of AGs or other drugs that are marketed according to the NDA[18].
This is for Lauren, my mother explained, smiling. The lady appeared not to have heard, and continued staring blankly at my mother. She hadn t noticed the bright bags at her feet. I quickly reached over and shut off the ignition. My mother got out and once again explained, We ve left something for Lauren - it s for Christmas. The lady s dark eyes softened11, and she smiled. She seemed too stunned12 for words. Offering a simple, Merry Christmas, we drove off leaving the woman still standing13 in her doorway14, smiling.
Subsequently,the Centers for Medicare &Medicaid Services (CMS) of HHS issued 72 FR 39141 rule document in 2007[19],which explained the specific rules of calculating AMP and BP of brand-name drugs including AG.And NDA holders must take into account the transfer price of AGs to secondary manufacturers when calculating the best price,thus making the difference between AMP and BP (usually including the transfer price of AG)greater.So,the refund discount amount of the brandname drug enterprise providing AG becomes larger.(See Fig.2).

Fig.2 AG and Medicaid drug rebate program and regulatory documents
Medicaid discounts are expected to increase for the majority of brand-name drug manufacturers following the release of the CMS rules document.The financial impact caused by the increase in discounts for brand-name drug manufacturers may also inf luence the corporate strategy of their marketed AG.Some companies may choose to abandon marketing AG due to the meagre prof its of AG and the increased Medicaid discounts.As a result,the Medicaid discount has become an important part to maintain the balance between innovative drugs and generic drugs.
5 Case studies—the legality of AG
The legality and policy debate of marketing authorized generic drugs has always been the focus of FDA and the US Congress.Different people hold diff erent opinions on it.The opponents of AG believe that the exclusive term of generic drugs in the “Hatch-Waxman Act” should be understood as not allowing the marketing of AGs within the 180-day exclusive period.While supporters believe that the Act does not require brand-name drug manufacturers to submit any kind of marketing application for AG,so the exclusive period is only applicable to ANDA or 505(b) (2)application with PIV certification,and AGs should not be prohibited from entering the market.
Disputes over the legality of AG’s marketing during the exclusive period have been heard in many cases.In Teva Pharmaceutical Co.,Ltd.vs.Crawford[20],the United States Court of Appeals for the Federal Circuit (CAFC) held that it was unreasonable to interpret the relevant provisions of the“Hatch-Waxman Act” as prohibiting the authorization of AGs to market during the 180-day exclusive period.In this case,independent generic drug manufacturer Teva has previously reached an agreement with PurePac Pharmaceutical Company (the first ANDA filer to submit PIV certification) to share the 180-day exclusive period for the drug gabapentin.It means PurePac Pharmaceutical Company agreed to distribute the first generic drug in exchange for part of its income during the exclusive period.On June 6,2005 (180-day exclusive period),Pfizer marketed its “generic”version of gabapentin (i.e.AG),which is much cheaper than that of the same brand-name drug (Neurontin),packaged as generic drug and sold through many of the same channels as Teva’s generic products.
Teva required FDA to prohibit the AG of gabapentin from marketing within 180-day exclusive period,and FDA shall require Pfizer to submit a supplementary new drug application (sNDA) when marketing or distributing any drug that changes the brand-name drug version to be similar to or easily confused with the generic drug according to 21 U.S.C.section 356a (b)[21].FDA rejected Teva’s request and concluded that it does not consider or refuse to delay the marketing of AG and further pointed that there is no legal basis to impose the application approval requirements on the marketing of AG.Teva filed a lawsuit against FDA,and the local court agreed with FDA’s view that nothing in the regulations supports the argument that FDA can prohibit NDA holders from marketing AG during the exclusive period.Teva then appealed to CAFC in Washington,D.C.
As shown in the “Hatch-Waxman Act”,Congress is trying to strike a balance between encouraging innovation and quickly marketing the low-cost generic drugs.To achieve a balance between the conflicting goals should be done by the law-making body,so the court must strictly abide by the terms of the judgment expressed by Congress.
Congress deemed it appropriate and prescribed the incentives for generic manufacturers to challenge brand-name drug patents.FDA shall not approve the second and subsequent ANDA with PIV certification until 180 days after the first generic filer starts commercial sales of the drug or wins a court decision against the patent holder.It is incomprehensible to require FDA to impose restrictions on AGs in accordance with the above regulations,because AG does not need to submit an ANDA application,and the NDA holders will not challenge their patents.Moreover,nothing in section 365 a (d) mentions that FDA requires an approved new drug applicant to submit a sNDA for a “non-material manufacturing change”.As pointed out by FDA,the purpose of submitting sNDA according to section 356 a (b) is to verify the influence of production changes on the characteristics,strength,quality,purity and efficacy of drugs,as they may be related to the safety or effectiveness of drugs.FDA will not require sNDA for reasons unrelated to the safety and effectiveness of brand-name drug.
Because there is no clear provision on this issue in the current laws,bills and other statutory laws of the US,and “Case Law” is the backbone of American law,the judicial judgments of Tevavs.Crawford case currently represent the legal support of this type of case.In the absence of further judicial interpretation or legislative action by Congress,AG will be considered as a legal means for NDA holders to market their products under the Hatch-Waxman Act[9].
6 The inspiration of AG system in the US to China
6.1 Analysis of the necessity of establishing AG system in China
Since 2017,many policies have been introduced at the national level,which explicitly propose to establish a drug patent-linkage system.With the official signing of the “Government Economic and Trade Agreement[22]” between China and the US on January 15,2020,it is inevitable to accelerate the introduction of drug patent-linkage system in China.At present,the government is actively constructing the drug patent-linkage system.First of all,the“Drug Administration Law” defines the drug patentlinkage system at the upper level,and then some clear regulations will be introduced.
Through the research,it is found that the AG system was not mentioned in various documents in China.In the US,AG system is the product of “Hatch-Waxman Act” and patent-linkage system,which plays an important role in the pharmaceutical market.Through the study of AG competitiveness,it is found that the “no AG” settlement has a certain function of anti-competitiveness and monopoly.If there are no legal restrictions on it,after the patent-linkage system is fully implemented in our country,it will inevitably lead to an increase in patent lawsuits.At the same time,AG has an incomparable first-mover advantage over independent generic drugs.If relevant marketing procedures and regulations are not formulated,brandname drug manufacturers marketing AG in China through its subsidiaries or foreign generic drug giants will undoubtedly crack down on domestic generic drug companies.
To sum up,patent linkage system is not an isolated system,and it can be effectively implemented only by establishing a supporting system,in which the construction and design of AG system is particularly important.
6.2 Establishing and optimizing patent-linkage system based on China’s national conditions
When introducing the patent-linkage system in China,it should be studied and improved in light of China’s national conditions.A complete patent-linkage system usually includes patent infringement,patent registration and information publicity,statement of patent ownership status,patent challenge and litigation,approval waiting period,generic exclusive period and authorized generic system,etc.
First,it is clear that patent infringement is the premise of establishing a drug patent-linkage system.Article 35 U.S.C 271 provides provision for patent infringement,i.e.filling 505 (j) or 505 (b) (2)ANDA application according to FD&CA,aiming to obtain approval for marketing,use or sale before the expiration of the patent,and the application shall be deemed as infringement of the patent right.At present,the China’s “Patent Law” does not stipulate that the act of submitting a marketing application by a generic drug applicant is regarded as infringement,thus the process of patent challenge cannot be started.Therefore,it is necessary to explicitly formulate patent infringement provisions in the “Patent Law”.Secondly,in order to avoid the unreasonable extension of patent caused by patent evergreen,we should stipulate the type of registered drug patent and the examination mechanism of the validity of registered patent.When submitting the application for marketing,the application of new drugs in China shall disclose the patent information required and publicize it in the catalogue of Chinese Drugs Listing.By examining registered patents,it is also possible to avoid triggering multiple approvals waiting periods.In addition,the first generic incentive system should be established,and the appropriate exclusive period should be set to ensure that the generic drug manufacturers have enough motivation to challenge patents.Meanwhile,the rules of losing exclusive period should be made to avoid the infinite delay of generic drug marketing.
Through CNKI’s search of Chinese database and related resources,it is found that scholars and experts have conducted in-depth analysis on the relevant supporting systems of patent-linkage system except for the AG system.Therefore,based on the study of the system in the US,some suggestions and analysis on the AG system are put forward.
6.3 Promoting the construction of supporting system for AG
AG strategy is closely related to pharmaceutical industry stakeholders,healthcare providers and patients.Moreover,AG will have some impacts on the strategies and decisions of brand-name drug manufacturers and generic drug manufacturers.At present,China is actively establishing patent linkage system,which aims to meet the needs of patent protection of brand-name drug manufacturers and accelerate the marketing of generic drugs.As a key supporting system,AG should be comprehensively analyzed and studied.Through the analysis and research of American system,the establishment and improvement of AG system in China need to clarify the following elements.(See Fig.3).
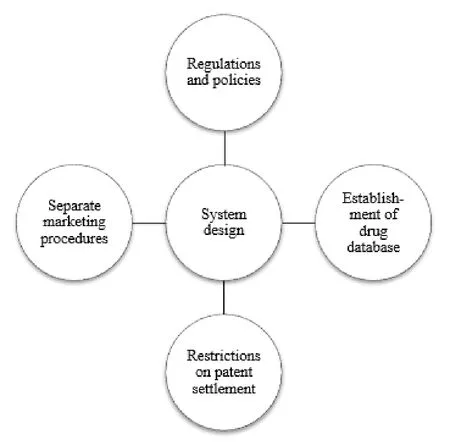
Fig.3 The elements of establishing AG system in China
6.3.1 Establishing regulations and policies on AGs
Research on AGs should be covered by the patent linkage system,and regulations and guidelines on AGs must be established.In the past 10 years,the global drug market entered a period of patent cliff.AGs have become a strategy for brand-name drug manufacturers to seize the market and maintain revenue.At present,China has the biggest market of generic drugs,and the market scale continues to expand.Brand-name drug manufacturers pay more attention to China’s generic drug market after the expiration of drug patents.However,China has not formulated relevant laws and policies on AGs.If the drug patent linkage system cannot include AG drug system,the goal of encouraging China’s generic drug companies to market in advance will be influenced by of foreign brand-name drug manufacturers’ AG strategy,which means they will face fierce competition in the market.
6.3.2 Establishing a separate procedure for AGs to enter the market
When establishing patent linkage system in China,it is necessary to clarify the relevant procedures for authorizing the approval and marketing of generic drugs.The marketing of AGs in the US only needs to be notified to FDA in the form of an annual report.Besides,the withdrawal does not need FDA approval.This filing procedure makes the information of AGs unclear.In order to better supervise AG,independent procedures for marketing and delisting of AGs should be made in China,and relevant information such as dosage forms,specifications,NDA applicants,distributors,and withdrawal applications should be submitted to the drug administration department.
6.3.3 Relevant drug databases should be established for AG
In the US,AG is not included in the Orange Book,but a separate AG list database has been established and updated quarterly.In China,National Medical Products Administration (NMPA) has formulated the “Catalogue of Chinese Listed Drugs”,which includes innovative drugs and improved new drugs with complete and standardized safety and effectiveness research data,as well as generic drugs with pharmaceutical equivalence and bioequivalence.Since the marketing of AG depends on the NDA application of brand-name drug manufacturers,its relevant information and drug essence are exactly the same as brand-name drugs,and it can cause drug confusion if AG is included in the Catalogue.Therefore,it is suggested to establish a separate database and carry out dynamic update management,so as to disclose the newly approved and withdrawn drugs in time.
6.3.4 Regulations on the patent settlements shall be formulated
The patent settlement of AG is widely used in the patent linkage system.The “Hatch-Waxman Act” does not specify the marketing of AG and whether it can be marketed during the exclusive period of generic drugs.There have been many lawsuits in the US on the legality of AG.Finally,the legal status of AG has been determined by the form of case law.Therefore,in order to avoid the occurrence of litigation,when issuing the corresponding laws,regulations and documents,it should be clearly stated whether it is allowed to market AG within the exclusive period of generic drugs.If AG is allowed to be marketed for sale during the exclusive period,regulations for the settlement shall be formulated,such as the limited time for “no AG” and the choice of AG distributors,etc.The settlement signed with the first filer and subsequent applicants shall also be reported to the drug administration and relevant departments for examining the competition and monopoly effects of the agreements.Meanwhile,whether the time for marketing independent generic drugs has been delayed and the influence on the consumers should be assessed.In addition,the interest of China’s generic drug companies can be maintained by limiting the inclusion of AG drugs in the medical insurance catalogue.

- 亚洲社会药学杂志的其它文章
- Research on the Construction of Pharmacy Organization and the Development of Pharmaceutical Services in China’s Primary Medical and Health Institutions— Based on the Survey of 5 Provinces
- DRG Payment System in the United States and Its Enlightenment to China
- Research on the World’s Most Valuable Antihypertensive Drugs Based on Patent Analysis
- Enlightenment and Reference of Vaccine Clinical Trial Design Based on Immunological Surrogate Endpoints in the United States
- Literature Review on the Development of New Drugs Based on TRIZ
- Research on the Relationship between Export Orientation and R&D lnvestment of China’s Pharmaceutical lndustry in Different Regions