
焙烧温度对Cs/SBA-15催化乙酸乙酯和甲醛羟醛缩合反应的影响
2021-03-17 07:06田恒水
天然气化工—C1化学与化工 2021年1期
田 翔,田恒水
(华东理工大学 化工学院,上海 200237)
丙烯酸及丙烯酸乙酯被广泛应用于涂料添加剂、粘合剂、纺织品和皮革处理剂等领域[1]。 目前丙烯酸主要由丙烯两步氧化法制取,再经过酯化反应制取丙烯酸乙酯[2]。 但是,我国“多煤少油缺气”的能源结构和不断扩张的石化产能不断推高原油的对外依存度,开发非石油路线生产丙烯酸(酯)显得十分迫切。 另外,乙酸和甲醛作为煤路线下游产品目前处于产能过剩的状态[3],气相羟醛缩合制备丙烯酸(酯)具有很强的经济潜力而备受关注[4]。
目前丙烯酸乙酯的羟醛缩合工艺尚处于起步阶段[5,6],酯类羟醛缩合的研究重点集中在对乙酸甲酯[7]和丙酸甲酯[8]的工艺开发。SBA-15具有比表面积大、孔径均一和酸性适中等优点,作为载体在制备丙烯酸[9]和甲基丙烯酸甲酯[10]的工作中被证明具有较优的催化性能。Si-O-Cs结构已被证明是羟醛缩合反应的活性位[11,12],而焙烧工艺作为催化剂制备的重要步骤,对羟醛缩合法制备丙烯酸乙酯工艺的影响还未见报道。
鉴于此,本工作在不同焙烧温度下初步探索硝酸铯在载体表面生成活性结构的过程,结合多种表征手段考察催化剂结构和酸碱性质的变化,进而测试在乙酸乙酯与甲醛羟醛缩合反应中的催化性能,并初步探讨酸碱性质对主副反应的影响。
1 实验部分
1.1 催化剂的制备
SBA-15的制备方法[13]:取10.67 g的P123和80 mL二次蒸馏水, 在1 L三口烧瓶中充分搅拌, 再加入320 mL的2 mol/L盐酸溶液, 并转入水浴锅中搅拌。随后,22.67 g正硅酸四乙酯在310 K下一次性加入,恒温搅拌24 h得到乳白色悬浊液,再移至500 mL水热釜中,在373 K下晶化处理48 h。 最后,经过滤、水洗和干燥处理所得到的白色粉末固体先在623 K马弗炉中保持2 h, 再升温至823 K焙烧6 h去除模板剂,制得SBA-15载体,标记为S。
Cs负载型催化剂的制备方法[14]:将0.0880 g的硝酸铯溶解在34.80 g二次蒸馏水中, 再加入1.2000 g载体,室温搅拌2 h后,在373 K烘箱过夜干燥,所得样品标记为Cs/S-D。 上述操作重复5次,样品混合备用。 Cs/S-D在通有空气的马弗炉分别于不同温度下焙烧5 h,分别标记为Cs/S(523)、Cs/S(623)、Cs/S(673)、Cs/S(723)和Cs/S(773)。
1.2 催化剂的表征方法
XRD采用Bruker D8 Advance型X射线衍射仪,辐射源为Cu Kα,管电流电压分别为40 mA和40 kV,以0.02°为步长分别在0.5°~6.0°和10°~60°范围进行扫描。
N2吸附/脱附采用Micromeritics 3 Flex 型分析仪,先在真空环境613 K下脱气6 h,随后在液氮温度下进行N2吸附测试。 用BET法计算样品的比表面积(SBET),对脱附支用BJH法估算孔体积(Vp)和平均孔径(dp)。
XPS使用Thermo Fisher公司的ESCALab-250Xi型多功能表面分析仪, 单色Al靶Kα射线为光源,以C 1s = 284.8 eV为内标。
吡 啶-FTIR 使 用Thermo Fisher Nicolet IS50 设备,样品在原位池真空673 K预处理2 h后加入吡啶,吸附平衡后用分子真空泵在373 K去除物理吸附的探针分子,降至室温后进行红外扫描。
NH3-TPD使用Micromeritics Autochem II 2920设备,取约50 mg样品在氦气氛围673 K下脱气1 h。 降至323 K后,通入NH3体积含量为10%的氦气,达到平衡后以氦气吹扫。 随后, 由热导检测器在323~823 K范围内对样品脱附的NH3进行测定,升温速率为10 K/min。
DTA/TGA测试采用SDT Q600型热重分析仪,空气氛围下对样品以10 K/min的升温速率在298~1298 K范围进行测试。
1.3 催化剂性能评价
催化剂性能评价在常压固定床反应器中进行,反应管的长度和内径分别为50 cm和0.8 cm并垂直放置于加热炉中, 在反应管中段填充0.3000 g催化剂。装置通入20 mL/min氮气并在663 K稳定后,将配比为n(乙醇):n(乙酸乙酯):n(甲醛)=4.0:2.5:1.0的原料通过平流泵以0.1 mL/min的流量输入反应器,其中三聚甲醛为甲醛来源。 反应经过1 h稳定后,收集随后2 h的反应产物,经过冷凝后的液体产物储存在收集罐中,尾气夹带的低沸点产物通过装有5 mL乙酸丁酯的吸收瓶进行收集。 将所得产物配制于乙酸丁酯和二甲基亚砜的混合液中, 甲苯为内标,在配有FID检测器和HP-5的毛细管柱的气相色谱上进行定量分析。 另外,液体产物加入含有双(三甲基硅烷基) 三氟乙酰胺的甲苯溶液中, 在333 K下进行30 min衍生化后, 以乙酸丁酯为内标, 对产物中的酸组分进行色谱定量分析。 本工作中的主产物为丙烯酸乙酯(EA)和丙烯酸(AA);副产物分别为乙醇脱氢反应生成的乙醛(ACE),及其下游产物丙烯醛(ACR)、乙酸甲酯(MeOAc)和丙酸(PA)。 以甲醛为基准计算各产物的收率(Y,%)、选择性(S,%)、空时收率(STY,mmol/(h·g))及物料平衡率(Bm,%):
式中,nI,OUT和nFA,IN分别为产物I的生成量和甲醛的输入量,mmol;nSUM,OUT为甲醛相关产物的总生成量,mmol;t为反应时间,h;mcat、mL、mG和mIN分别为催化剂质量、收集罐液体产物质量、尾气吸收瓶中轻组分产物质量和原料总输入质量,g。
2 结果与讨论
2.1 催化剂的表征
2.1.1 催化剂的结构性质
各样品的XRD谱图如图1所示。 在图1(A)中,所有样品均清晰地呈现出三个分别对应p6mm空间群的(100)、(110)和(200)晶面的特征峰[13],说明载体SBA-15的规整结构在经过各步处理后可得到良好保持。 图1(B)中,除2θ = 23°非定形二氧化硅的衍射峰外,在19.86°、28.28°、34.84°、40.38°、45.40°和50.08°处检测出对应硝酸铯(PDF 09-0403)的特征衍射峰,以星号标记。 硝酸铯的特征峰信号在523~673 K范围逐渐减弱,直到高于723 K后基本消失。
图2(A)为各样品的N2吸附/脱附等温线,根据IUPAC分类法, 均为带有H1型滞回环的IV型等温线,证明各样品的平行圆柱形孔道无明显堵塞[15]。如图2(B)所示,各样品的孔径分布变化不大,说明孔道结构保持了良好的均一性。
XRD和N2吸附/脱附方法测试的样品结构参数列于表1。 与S相比,Cs/S-D迅速增大的晶面间距(d100)和晶胞参数(α0)说明,SBA-15的六边形蜂窝状结构的边长和相邻孔道的中心距离同时增大,证明经过浸渍干燥处理后,载体的硅骨架发生一定程度的晶格膨胀。 变化不大的总比表面积(SBET)和孔容(Vp)可以说明在浸渍干燥过程中,硝酸铯与孔壁之间不会发生明显的化学作用。这可归因于Cs+较大的离子半径和较弱的孔壁渗透能力[12]。
样品Cs/S-D经不同焙烧温度处理得到Cs/S(523)至Cs/S(773)。 图1(B)已说明,随着焙烧温度升高,分布在载体表面的硝酸铯逐渐分解。 样品α0和壁厚(twall)随焙烧温度升高逐渐变小,结合逐渐变大的孔径(dp)可以说明,随着硝酸铯的分解,Cs+在生成活性位的过程中,不仅消耗了孔壁表面的硅材料,还引起了硅骨架的收缩。 这种因负载金属对硅骨架产生收缩的现象也出现在SBA-15负载Zn[16]、Cu[17]和Cs[11]等案例中。 迅速降低的SBET和Vp则说明,催化剂的传质条件随焙烧温度升高而变差。
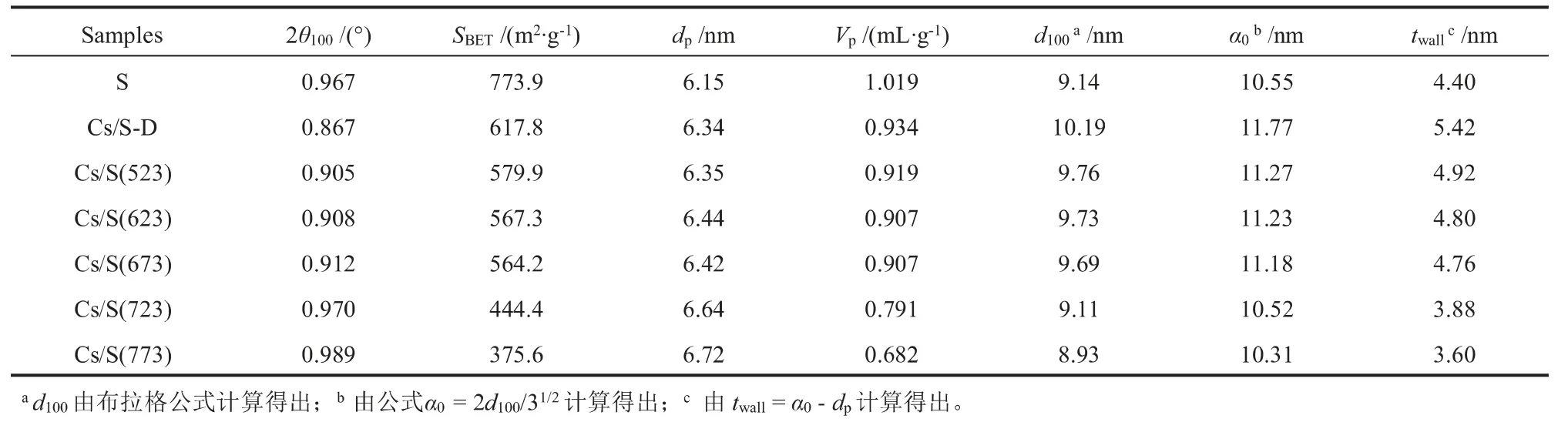
表1 各样品的结构参数
2.1.2 催化剂的酸碱性质
焙烧温度对催化剂碱性质的影响,通过各样品XPS的O 1s和Si 2p结合能谱进行了考察。Si-O-Cs作为活性结构,Cs通过电子效应增强了晶格氧的电负性,从而使其具备碱性功能[18]。 这种电子诱导效应还会传递到相邻的硅原子,使其结合能向低能方向迁移[19]。母体SBA-15的O 1s和Si 2p分别为533.10和103.75 eV,与文献相符[12]。另外,Si-O-Cs结构中的O 1s结合能为532.0 eV[14],NO3-的O 1s结合能为533.0 eV[20],均略低于载体。
样品Cs/S-D的Si 2p结合能与母体SBA-15基本一致,而硝酸根的引入使O 1s向低能方向产生了非常微弱的迁移,说明在浸渍干燥过程中不会生成活性结构。 随着焙烧温度从523 K上升至773 K,硝酸铯的残留量逐步降低,O 1s与Si 2p向低能区的迁移逐渐明显,则说明硝酸铯的分解与活性位的生成是同步发生的关系,且催化剂的碱性逐渐增强。 但是,与Cs/S(723)相比,Cs/S(773)的Si 2p继续向低能区迁移,而O 1s反而出现增大的现象,说明部分Cs+与硅材料生成了除Si-O-Cs以外的组成,如硅酸盐这类碱性更弱的物质[21]。
通过吡啶-FTIR和NH3-TPD考察了焙烧温度对催化剂酸性质的影响,结果如图4所示。图4(A)中,所有样品均只在1596和1445 cm-1处呈现出对应弱Lewis酸的表面硅羟基的特征峰[22],其信号强度在523 K以下变化并不显著, 当焙烧温度高于623 K后迅速减弱。图4(B)中各样品均在表示弱酸性的330~450 K范围出峰, 其峰面积的变化趋势与吡啶-FTIR的结果一致。 结合XPS结果可知,表面活性位由CsNO3与表面硅羟基的反应所形成,而催化剂的弱酸性主要来自残留的硅羟基, 其酸量随焙烧温度升高而逐渐减少。
2.1.3 热重分析
图5为Cs/S-D的DTG-TGA曲线。 温度低于390 K时,约有3%的质量损失,对应着物理吸附的水分。第二个失重平台出现在400~843 K范围,约9%,DTA曲线仅有一个失重峰, 最大失重速率对应的温度为684 K,记为Td-max。 此阶段的失重主要源于硝酸铯的分解和表面硅羟基的损失。当焙烧温度高于833 K时,失重趋向于0,可认为前驱体已完全分解且不再消耗硅物质。 第二失重阶段按失重速率可分为400~600 K的慢速区和600~780 K的快速区。 由此可见,焙烧温度决定了硝酸铯分解以及与硅基材料生成活性位Si-O-Cs的能力,进而影响催化剂的酸碱性质。
综合上述表征结果可知, 当温度低于Td-max时,硝酸铯与硅基材料的反应活性较弱,生成的活性位的数量较少;当温度高于Td-max时,随着前驱体与硅材料反应的增强,活性位数量逐渐增多而增强了催化剂的碱性,同时表面硅羟基的残留量也逐渐减少使酸性变弱。 但是,过高的焙烧温度会导致弱碱性物质的生成而使碱性减弱,并一定程度上降低了传质条件。
2.2 催化性能评价
不同焙烧温度制备的Cs/SBA-15在乙酸乙酯与甲醛的羟醛缩合反应的测试结果如表2所示。 当焙烧温度低于723 K时,随着焙烧温度升高,主产物的总空时收率、选择性和收率逐渐升高,而ACE及其下游产物则出现明显下降的趋势。 结合酸碱性质的表征结果可知,更多活性位的生成增强了催化剂碱性而有利于主反应的进行, 而表面硅羟基作为弱Lewis酸,其残留量对ACE的空时收率展示出较大影响。 实际上,相对更强的Lewis酸,如ZrO2[23]和MgO[24],已被证明有利于乙醇脱氢反应。 从表中各产物选择性的变化趋势可知,乙醇脱氢反应对主反应具有一定的抑制作用。 具体来说,ACE因具有α氢结构,会与乙酸乙酯竞争甲醛以发生羟醛缩合反应,而生成MeOAc的Tischenko反应[25]则会进一步消耗甲醛。 另外,PA的存在说明主产物AA还会发生加氢反应而损失。 因此,适当提高焙烧温度不仅有利于生成更多的活性位而提高主产物的收率,还有利于抑制乙醇脱氢反应而降低对主反应的负面影响。
但是,当焙烧温度进一步提高至773 K时,部分硝酸铯与表面硅羟基生成弱碱性组分导致碱性变弱,这应该是Cs/S(773)主产物总空时收率和收率下降的主要原因。 另外,物料平衡率和表1中各结构参数的变化趋势似乎可以说明,变差的传质条件会一定程度上导致部分产物损失而降低主产物的收率。
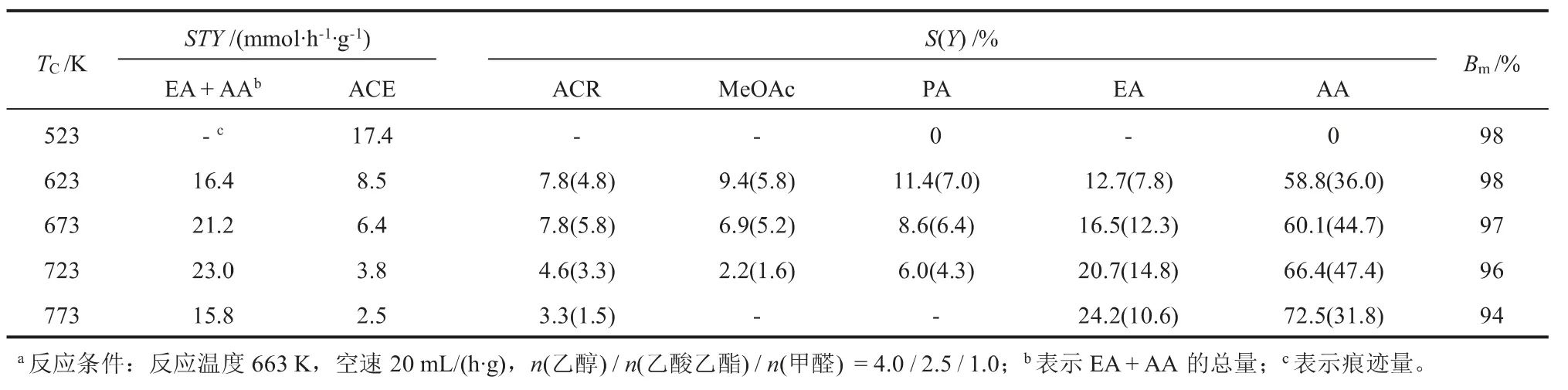
表2 不同焙烧温度处理Cs/SBA-15的性能测试a
3 结论
本工作通过不同焙烧温度制备了Cs/SBA-15催化剂,用于乙酸乙酯与甲醛羟醛缩合制备丙烯酸乙酯和丙烯酸的工艺中。 强碱弱酸的酸碱性质,不仅有利于主反应,还有利于抑制副反应乙醇脱氢的发生。 723 K焙烧制备的催化剂具有较佳的反应效果,丙烯酸乙酯和丙烯酸的总收率和总选择性分别为62.2%和87.1%,空时收率为23.0 mmol/(h·g)。研究结果为认识主副反应之间的关系和Cs负载型硅基催化剂的深入开发提供了依据。
猜你喜欢
化工管理(2022年24期)2022-11-09
云南化工(2022年2期)2022-03-18
农药科学与管理(2021年2期)2021-03-16
作文周刊·八年级版(2020年8期)2020-05-25
黄河黄土黄种人(2019年11期)2019-12-20
阅读与作文(英语高中版)(2019年8期)2019-08-27
健康博览(2019年1期)2019-04-08
大陆桥视野·下(2017年12期)2017-11-29
教育(2016年49期)2017-03-20
