
他唑巴坦杂质A国家标准物质的研制
2021-03-13 06:53:52马步芳刘书妤姚尚辰冯艳春
中国药科大学学报 2021年1期
肖 亭,王 晨,田 冶,张 夏,刘 颖,马步芳,刘书妤,姚尚辰*,冯艳春**
(1中国食品药品检定研究院,北京102629;2上海工程技术大学,上海201620)
国家药品标准物质系指供国家法定药品标准中药品的物理、化学及生物学等测试用,具有确定的特性或量值,用于校准设备、评价测量方法、给供试药品赋值或鉴别用的物质,应具备稳定性、均匀性和准确性[1]。标准物质的制备是药品质量研究中最重要的一环。近年来,随着公众健康安全意识和分析技术手段的不断提高,标准中对于杂质的定向控制也越来越细化。使用杂质标准物质对质量标准中要求的杂质进行定性和定量的控制是最理想的杂质控制方法。因此无论从监管理念的转变、药品质量的提升还是市场需求等多个角度,杂质标准物质的研制逐渐成为标准物质研制的发展方向[2]。
目前在各国药典中,仍有很多采用原位降解技术[3]或利用相对保留时间定位来进行某些杂质的控制或用于检查方法的系统适用性。但原位降解技术过程繁琐、产生杂质有限且重现性较差,不同实验室由于实验条件的差异可能造成色谱峰相对保留时间的改变[4],使得许多药典方法在执行过程中遇到困难。因此,在条件许可的情况下逐渐将原位降解技术产生的杂质逐一研制成杂质标准物质或者进行混合杂质标准物质的研制[5]成为药典标准修订中一项重要的工作。
他唑巴坦(tazobactam)是从舒巴坦的衍生物中筛选出来的一种β-内酰胺酶抑制剂。它可以与数种β-内酰胺类抗生素产生协同作用,增加了β-内酰胺类抗生素的抗菌活性并扩大了其抗菌谱[6]。因此《中华人民共和国药典》(以下简称“中国药典”)自2010 版收录了该品种原料的质量标准,其中有关物质检查项和含量测定项都使用了他唑巴坦的氢氧化钠降解溶液作为系统适用性溶液,使用降解产生的杂质来判断系统的分离效果,该方法一直沿用至今。本研究以中国药典(2015 年版)二部中他唑巴坦原料药质量标准为例,介绍如何鉴定其含量测定和有关物质检查方法中通过原位降解技术获得的系统适用性试验溶液中的主要杂质,并按照杂质标准物质研制的一般流程[7],将其合成并研制成为国家杂质标准物质。
1 材 料
1.1 样品与试剂
他唑巴坦杂质A 原料(100 g,色谱纯度98%,河北桑迪亚医药技术有限责任公司);他唑巴坦对照品(99.5%,批号130511-201904,中国食品药品检定研究院);他唑巴坦杂质A 对照品(定性使用,R089H0,美国药典委员会);乙腈、甲醇(色谱纯,美国Fisher公司),其他试剂均为市售分析纯,水为超纯水。
1.2 仪 器
Waters 2695 Alliance 液相色谱仪(美国Waters公司);QTrap6500 质谱仪(美国Sciex 公司);FT-IR EQUINOX 55 型红外光谱仪、Ascend 500 核磁共振仪(德国Bruker 公司);V20 型水分滴定仪(美国Mettler 公司);FO410C 型马弗炉(日本Yamato公司)。
2 方 法
2.1 中国药典(2015 年版)他唑巴坦原料药有关物质检查方法系统适用性试验溶液中最大杂质的鉴别
根据中国药典(2015 年版)二部中他唑巴坦原料药有关物质检查方法所描述的原位降解方法制备系统适用性溶液:取他唑巴坦对照品约25 mg至25 mL 量瓶中,加0.01 mol/L 氢氧化钠溶液10 mL,30 ℃放置30 min,用0.01 mol/L 盐酸溶液中和后,加流动相稀释至刻度,摇匀,用磷酸调节pH 至4.0,按照含量测定色谱条件要求[色谱柱:Dikma,Inertsil ODS-2(4.6 mm × 250 mm,5 μm);C/N 5020-01128;S/N 8AS11013;流 动 相:乙 腈-0.03 mol/L 磷酸二氢钾溶液-10%四丁基氢氧化铵溶液(190∶795∶15)(用磷酸调节pH 至4.0);流速:1.0 mL/min;检测波长:230 nm,进样20 μL,进行HPLC色谱分析,观察产生的主要杂质。
根据产生的主要杂质,采用液相质谱联用技术测定杂质的相对分子质量,推测其可能的结构。25 mL 量瓶内他唑巴坦的碱降解溶液用盐酸溶液中和后,用乙腈-水(1∶5)的溶液稀释至刻度。设计的色谱条件如下。色谱柱:Capcell PAK C18TYPE MGⅡ(4.6 mm×150 mm,5 μm);检测波长:230 nm;流动相A 相:0.1%甲酸水溶液,B 相:乙腈;流速:0.5 mL/min;梯度洗脱:0~0.01 min(95∶5);0.01~5.00 min(95∶5);5.00~17.00 min(38∶62);17.00~17.10 min(95∶5);17.10~22.00 min(95∶5)。
质谱仪型号为6500QTrap;工作站为Analyst®软件(Ver.1.6.2)。离子源:ESI,正离子模式。一级质谱图(Q1 scan)扫描的质量范围是50~500。对准分子离子母离子进行Enhanced Product Ion(EPI)扫描,得到二级质谱图。
2.2 杂质的合成
根据确定的结构式,设计合成路线进行主要杂质的合成。
2.3 杂质标准物质的研制
2.3.1 结构分析
(1)质谱分析 取他唑巴坦杂质A 原料适量,以乙腈-水(1∶5)为溶剂溶解并稀释成约0.5 mg/mL的溶液,用于质谱分析。质谱仪型号为6500QTrap;工作站为Analyst®软件。离子源:ESI,正离子模式。一级质谱图(Q1 scan)扫描的质量范围是50~500。对准分子离子母离子进行EPI 扫描,得到二级质谱图。根据准分子离子峰[M+H]+判断相对分子质量。
(2)红外分析 取他唑巴坦杂质A 原料约1 mg,置玛瑙研钵中与约干燥KBr 200 mg混合,缓缓研磨至细,压片。采用红外光谱仪测定各杂质的红外光谱,工作站为OPUS 光谱分析软件(Ver.5.5)。
(3)核磁共振波谱分析 取他唑巴坦杂质A约10 mg置于石英核磁管中,用D2O 0.5 mL为溶剂溶解,作为供试品溶液。采用配有CryoProbe Progdigy探头的核磁共振仪对杂质原料分别做1H NMR谱、13C NMR 谱、DEPT 90°谱、DEPT 135°谱、1H-1H COSY谱、HSQC谱和HMBC谱。
2.3.2 均匀性分析 从制备的2 900 瓶样品中随机抽取12 瓶样品,每瓶称取2 份,每份质量约为10 mg,采用纯度分析的液相色谱条件,以峰面积与对应称样量之比为评价指标,通过瓶间方差和瓶内方差的比较以判断样品的均匀性。如果两者之比小于95%置信水平对应F检验的临界值,则认为样品是均匀的。
2.3.3 稳定性分析 考察样品在实验色谱条件下溶液的稳定性,为确定样品的具体实验方案提供依据。目前标准物质原料短期稳定性考察主要是为样品选择合适的运输条件提供依据,为节约运输成本,首先考察了样品在常温(25 ℃)条件下0、3、5和10 d的稳定性,每个实验条件考察3支独立样品。
2.3.4 对照品赋值分析
(1)纯度分析 目前美国药典(USP41-NF36)、中国药典二部(2015 年版)和日本药典(JP17)均收载了他唑巴坦原料的质量标准,USP 与JP 色谱条件基本一致。实验中发现他唑巴坦杂质A 在酸性溶液中不稳定,容易降解,而中国药典和美国药典他唑巴坦有关物质检查方法中流动相和样品溶剂均偏酸性,因此在其纯度分析时未采用上述药典方法,而是参考文献[8],最终采用10 mmol/L 醋酸铵水溶液-乙腈(98∶2)为流动相和溶剂,色谱柱采用DIKMA,Inertsil ODS-2(4.6 mm×250 mm,5 μm)。采用Waters 2695 Alliance液相色谱仪。
(2)水分分析 精密称取杂质原料约30 mg,采用滴定仪,按中国药典四部(2015 年版)通则0832 水分测定法第一法(费休氏法)测定杂质的水分含量,溶剂为甲醇。
(3)无机杂质分析 炽灼残渣含量反映样品中无机杂质的量,采用中国药典四部(2015 年版)通则0841炽灼残渣检查法测定。
(4)对照品赋值 采用质量平衡法对对照品进行赋值。质量平衡法,即测得的活性成分的含量加上有机杂质、水分、残留溶剂、无机杂质的含量之和应为100%。计算公式:含量(%)=(1-有机杂质的质量分数)×(1-水的质量分数-挥发性物质的质量分数-无机杂质的质量分数)×100。
采用zg30 脉冲序列测定样品溶液的1H NMR图谱,测定温度298 K,观测频率400.13 MHz,90°脉冲宽度11.07 μs,扫描次数16 次,延迟时间10 s。以样品化学位移δ=7.794的信号锋为定量峰,顺丁烯二酸为内标(δ=5.907),测定待测物的含量,对质量平衡法测定结果进行确证。
3 结 果
3.1 中国药典(2015 年版)他唑巴坦原料药有关物质检查方法系统适用性试验溶液中最大杂质的鉴别
按照中国药典二部中他唑巴坦原料药有关物质检查方法规定的原位降解要求,制备系统适用性试验溶液,其色谱图如图1所示。在相对他唑巴坦峰保留时间0.4左右的位置出现一个较大杂质,目前中国药典标准要求他唑巴坦峰与其之间的分离度应不小于10。文献[9]记载他唑巴坦在体内会部分代谢为一种没有活性的产物,结构如图2,该代谢产物也可以通过NaOH 水溶液降解产生,另外他唑巴坦粉末或者冻干粉末也会热降解产生该杂质[10]。经检索,USP41 已经收载了该杂质,命名为他唑巴坦有关物质A(tazobactam related compound A,分子式:C7H12N4O4S;CAS:118175-11-4)。由此推测,中国药典(2015 年版)中他唑巴坦有关物质系统适用性试验溶液所产生的最大杂质可能为USP41 收录的他唑巴坦有关物质A。于是购买USP该对照品进行确认,经比对发现最大杂质与他唑巴坦有关物质A 有相同的紫外末端吸收、HPLC保留时间基本一致,质谱数据显示两者均出现249的[M+H]+峰,初步确认两者为同一物质。
3.2 最大杂质原料的合成
根据文献[8]记载的方法,按路线1 合成他唑巴坦杂质A原料100 g,纯度大于98%。
3.3 他唑巴坦杂质A结构确证
分别采用红外、质谱和核磁共振波谱对合成的他唑巴坦杂质A的结构进行了确证。
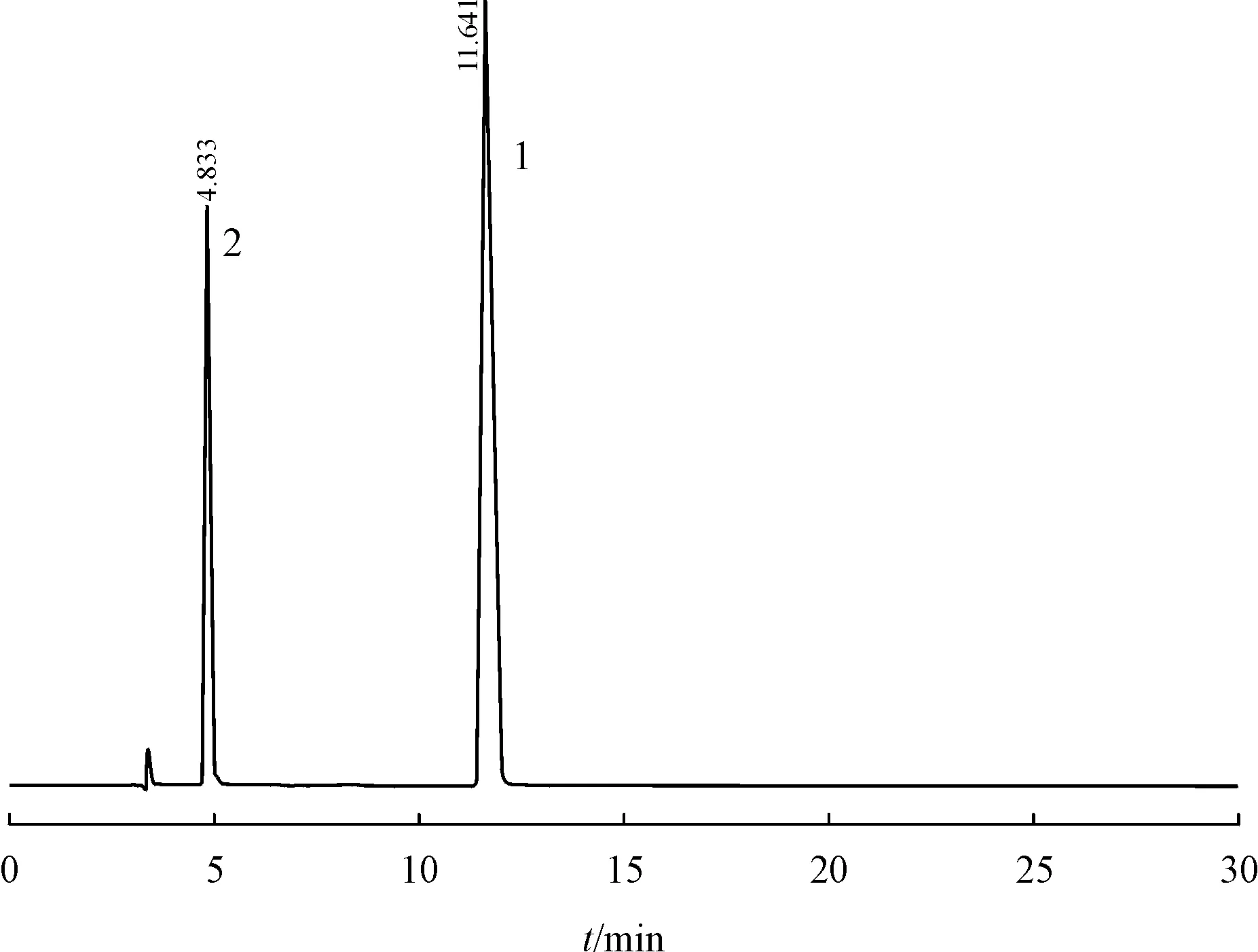
Figure 1 Chromatogram of the system applicability solution under the inspection item of tazobactam API related substances in Part 2 of Chinese Pharmacopoeia 2015
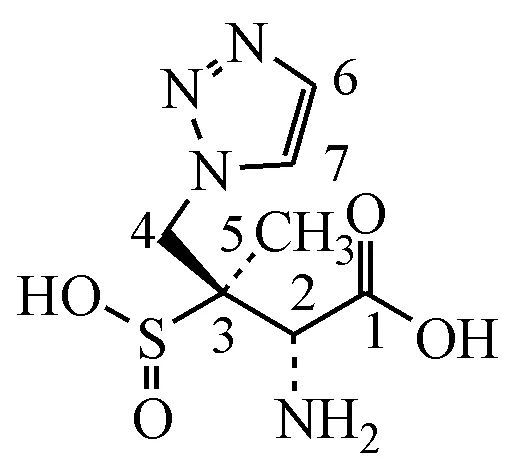
Figure 2 Structure of tazobactam impurity A
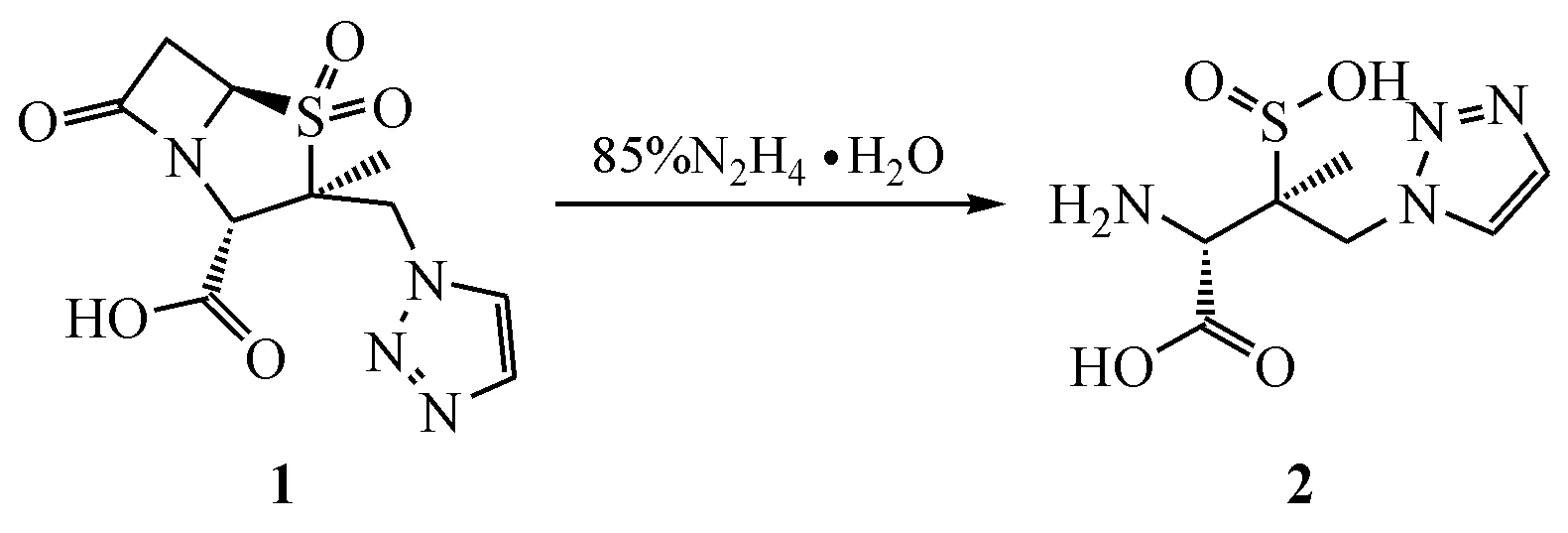
Scheme 1 Synthetic route of tazobactam impurity A
3.3.1 质谱推断 合成的他唑巴坦杂质A 原料电喷雾电离质谱(ESI-MS)显示他唑巴坦杂质A 与H+形成准分子离子[M+H]+,质荷比(m/z)为249.0,与其理论相对分子质量248.1相符合。
3.3.2 红外确证 红外光谱(图3)显示,合成的他唑巴坦杂质A 原料与USP 他唑巴坦有关物质A标准物质红外图谱基本一致。
3.3.3 核磁共振波谱确证 通过核磁共振波谱进一步确证合成的他唑巴坦杂质A 原料的结构。他唑巴坦杂质A 原料的氢谱、1H-1H COSY 谱、碳谱、DEPT(90°/135°)谱和HMBC 谱数据及解析详见表1 和表2。他唑巴坦杂质A 原料氢谱数据与文献[11]报道基本一致。通过解析可知,他唑巴坦杂质A 原料1H NMR 谱和13C NMR 谱中各峰的化学位移值与其化学结构式:(2S,3S)-2-氨基-3-甲基-3-亚磺酸基-4-(1H-1,2,3-三氮唑-1-基)-丁酸相吻合,所有峰均能在结构式中找到明确合理的指认,与USP 他唑巴坦有关物质A 对照品基本一致。
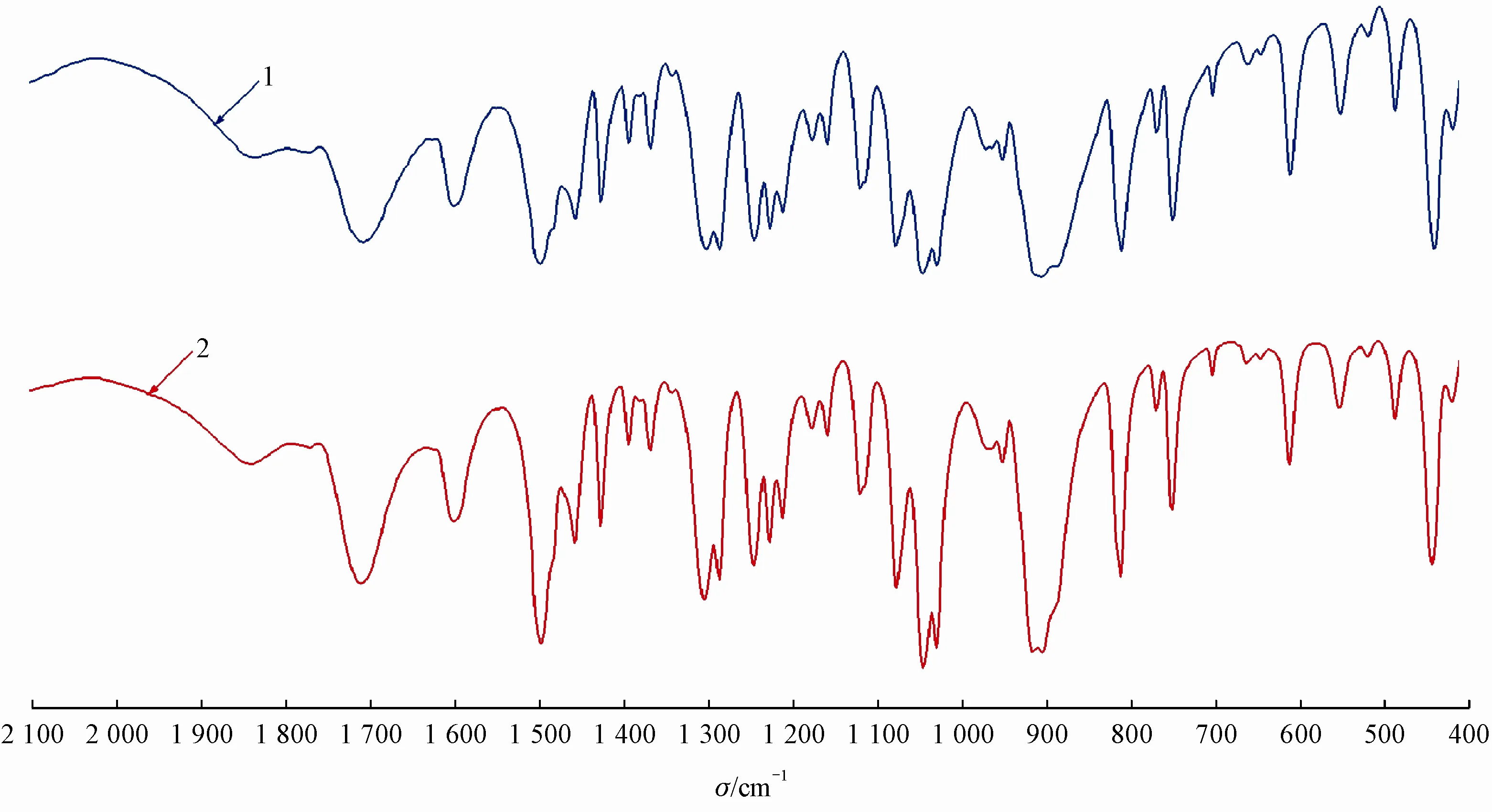
Figure 3 Infrared absorption spectrum of tazobactam impurity A and tazobactam related compound A contained in USP
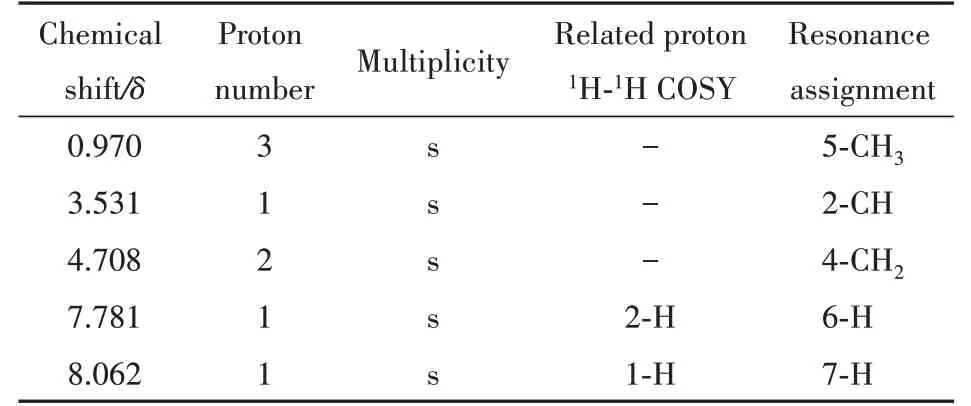
Table 1 1H NMR(D2O)data analysis for tazobactam impurity A

Table 2 13C NMR(D2O)data analysis for tazobactam impurity A
3.4 他唑巴坦杂质A原料均匀性和稳定性分析
对均匀性评价指标——24 份峰面积与对应称样量之比做F检验,在95%置信范围内,瓶间和瓶内F为0.61,小于F0.05(11,12)(2.79),证明瓶间与瓶内不存在显著性差异,该原料的均匀性良好。
实验中发现,采用中国药典二部(2015 年版)他唑巴坦原料有关物质检查的色谱条件,平行实验5份样品,他唑巴坦杂质A 面积归一化纯度均值仅为88.1%(RSD=1.96%),与合成公司交付色谱纯度(>98%)存在较大差异,怀疑样品发生降解。于是结合文献[8]重新摸索样品色谱纯度测定条件,最终采用10 mmol/L醋酸铵水溶液-乙腈(98∶2)为流动相和溶剂,等度洗脱,230 nm 为检测波长,测定不同稳定性考察的样品。以他唑巴坦杂质A面积归一化纯度为评价指标,观察不同时间结果的变化趋势。结果显示同一样品溶液在8 h 内的5 次进样,230 nm 处结果面积归一化纯度均值为99.1%(RSD=0.14%),且在紫外全波长范围内210~400 nm 未检出明显杂质峰,证明样品在该溶液中稳定。短期稳定性实验结果显示他唑巴坦杂质A 面积归一化纯度在0、3、5 和10 d 的均值均为99.1%,未发现降解趋势,证明样品在室温下10 d内稳定。对于样品的长期稳定性数据需要根据保存时间逐步考察并补充完善。
3.5 他唑巴坦杂质A原料赋值
采用质量平衡法计算他唑巴坦杂质A 原料的含量。首先需要进行纯度分析,确定所含有机杂质的多少。另外还需要分别确定水分、残留溶剂和无机杂质。根据原料的合成路线排除了残留有机溶剂的可能,因此仅使用残留溶剂数据库[11]对未知有机溶剂进行初筛,未检测到任何影响定量的有机溶剂峰。最后由有机杂质0.90%、水分1.24%和无机杂质(炽灼残渣)0.25%,计算出他唑巴坦杂质A原料的含量为97.6%,与核磁共振波谱定量结果97.1%基本一致。
4 结 论
本研究对中国药典(2015 年版)他唑巴坦质量标准中含量和有关物质检查项使用的原位降解方法得到的系统适用性溶液进行成分分析,确定了其中主要杂质的结构,并根据结构合成制备了该杂质标准物质,若使用该标准物质与他唑巴坦直接配置系统适用性溶液将使得标准操作更加简便易行,同时文献也检索到该杂质为他唑巴坦的降解产物,实验证明其在酸性环境下不稳定,则该标准物质的研制也为下一步提高他唑巴坦质量标准奠定了基础。
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28 06:26:50
中国感染与化疗杂志(2021年2期)2021-03-25 15:59:12
中华养生保健(2020年1期)2020-11-16 00:47:36
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
童话世界(2017年29期)2017-12-16 07:59:32
赤子(2017年4期)2017-06-30 04:56:05
北方牧业(2016年1期)2016-12-17 19:08:50
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
中国继续医学教育(2015年5期)2016-01-07 07:38:27
应用化工(2014年11期)2014-08-16 15:59:13
