
蜜蜂球囊菌菌丝和孢子中长链非编码RNA的比较及潜在功能分析
2021-03-08 08:37陈华枝王杰祝智威蒋海宾范元婵范小雪万洁琦卢家轩郑燕珍付中民徐国钧陈大福郭睿
中国农业科学 2021年2期
陈华枝,王杰,祝智威,蒋海宾,范元婵,范小雪,万洁琦,卢家轩,郑燕珍,付中民,2,3,徐国钧,陈大福,2,3,郭睿,2,3
蜜蜂球囊菌菌丝和孢子中长链非编码RNA的比较及潜在功能分析
陈华枝1,王杰1,祝智威1,蒋海宾1,范元婵1,范小雪1,万洁琦1,卢家轩1,郑燕珍1,付中民1,2,3,徐国钧1,陈大福1,2,3,郭睿1,2,3
1福建农林大学动物科学学院(蜂学学院),福州 350002;2福建农林大学蜂疗研究所,福州 350002;3福建农林大学蜂产品加工与应用教育部工程研究中心,福州 350002
蜜蜂球囊菌(,简称球囊菌)是一种专性侵染蜜蜂幼虫的真菌病原,可引起成年蜜蜂数量和蜂群群势的急剧下降。长链非编码RNA(long non-coding RNA,lncRNA)是一类新近发现的非编码RNA,在表观遗传、细胞周期、剂量补偿等众多生命活动中发挥重要生物学功能。【】明确球囊菌菌丝和孢子中lncRNA的数量、种类和表达谱差异,并探究共有lncRNA、特有lncRNA和差异表达lncRNA(differentially expressed lncRNA,DElncRNA)在菌丝与孢子中的潜在功能。利用基于链特异性建库的lncRNA-seq技术对球囊菌的纯化菌丝(AaM)和纯化孢子(AaS)分别进行测序。根据FPKM(Fragment Per Kilobase of per Million mapped reads)法计算lncRNA在AaM和AaS中的表达水平。通过Venn分析筛选AaM与AaS的共有lncRNA和特有lncRNA。按照≤0.05且|log2fold change|≥1的标准筛选AaM vs AaS比较组中的DElncRNA。通过Blast工具将共有lncRNA、特有lncRNA和DElncRNA的上下游基因比对GO和KEGG数据库,以进行功能及通路注释。根据靶向结合关系构建共有lncRNA、特有lncRNA和DElncRNA的竞争性内源RNA(competing endogenous RNA,ceRNA)调控网络并利用Cytoscape软件进行可视化。利用RT-qPCR验证测序数据的可靠性。AaM和AaS分别测得108 614 646和105 675 408条原始读段(raw reads),经严格过滤得到107 780 032和104 621 402条有效读段(clean reads),20分别为98.76%和98.72%,30分别达到95.84%和95.78%。共鉴定到850个lncRNA。AaM和AaS的共有lncRNA为701个,二者的特有lncRNA分别为39和110个。上述共有lncRNA通过顺式作用调控3 992个上下游基因,它们涉及细胞进程、代谢进程和催化活性等42个功能条目,以及代谢途径、次生代谢物的生物合成和抗生素的生物合成等117条通路;AaM的特有lncRNA和AaS的特有lncRNA通过顺式作用分别调控243和672个上下游基因。AaM vs AaS比较组包含的255个DElncRNA通过顺式作用调控1 479个上下游基因,它们涉及代谢进程、细胞进程和催化活性等41个功能条目,以及代谢途径、次生代谢产物的生物合成和抗生素的生物合成等107条通路。从共有lncRNA、孢子特有lncRNA和DElncRNA中分别预测到41、5和13个微小RNA(microRNA,miRNA)的前体序列。调控网络分析结果显示,菌丝lncRNA、孢子lncRNA形成较为复杂的ceRNA调控网络;菌丝lncRNA可靶向结合8个miRNA,进而调控77个mRNA;孢子lncRNA可靶向结合7个miRNA,进而调控87个mRNA;2个DElncRNA(TCONS_00008630与TCONS_00009302)可靶向结合miR-4968-y,进而调控10个mRNA。RT-qPCR验证结果显示4个DElncRNA的差异表达趋势与测序结果一致,表明测序数据真实可靠。共有lncRNA、特有lncRNA和DElncRNA可能通过调控上下游基因的表达,作为miRNA的前体,以及充当ceRNA影响菌丝和孢子的物质和能量代谢、自噬、转录、MAPK信号通路、泛素介导的蛋白水解、蛋白酶体以及次生代谢产物的生物合成等生物学过程,从而调节球囊菌的生长、发育、生殖和致病性。
蜜蜂球囊菌;长链非编码RNA;菌丝;孢子;竞争性内源RNA
0 引言
【研究意义】蜜蜂球囊菌(,简称球囊菌)是一种能够侵染蜜蜂幼虫而导致白垩病的真菌病原[1]。哺育蜂将被球囊菌孢子污染的花粉饲喂幼虫是导致该病的直接原因[2]。白垩病一般发生于春季和初夏时期,患病蜂群中的幼虫会出现成批死亡的症状,虽然该病不会导致蜂群的灭亡,但能导致成蜂数量和蜂群生产力的急剧降低,同时会使其他病原微生物入侵的概率增加,严重影响蜜蜂健康和养蜂生产[3]。然而,由于球囊菌菌丝生长、孢子形成和萌发的相关分子机理尚未阐明,蜜蜂白垩病的防治成效有限。利用基于链特异性建库的lncRNA-seq技术对球囊菌的孢子和菌丝分别进行测序和组学分析,能够明确球囊菌孢子和菌丝长链非编码RNA(long non-coding RNA,lncRNA)的数量、种类和表达谱差异,解析lncRNA介导的菌丝生长、孢子萌发、杂交产孢和毒力因子合成的生物学过程,为阐明相关分子机制提供重要信息和数据基础。【前人研究进展】lncRNA指的是一类长度>200 nt、含有2个及以上外显子且不具有蛋白编码能力的非编码RNA(non-coding RNA,ncRNA),多由RNA聚合酶II转录形成,具有类似于mRNA的结构如5′帽子和3′ polyA尾巴[4]。LncRNA曾经被认为是基因转录过程中的“暗物质”和“噪声”,但目前已被较多的研究证实能够作为信号[5]、诱饵[6]、引导[7]及支架分子[8],在表观遗传[9]、细胞周期调控[10]以及剂量补偿[5]等诸多生命活动中发挥关键的调控功能。根据lncRNA基因在基因组上与蛋白编码基因的相对位置,可将其分为基因间区lncRNA(intergenic lncRNA)、双向lncRNA(bidirectional lncRNA)、内含子RNA(intronic lncRNA)、反义链RNA(antisense lncRNA)和正义链RNA(sense lncRNA)[11]。LncRNA具有高度的物种、组织表达和发育时期表达特异性[12]。LncRNA与相邻蛋白编码基因的顺式()作用元件或3′UTR区域结合,从而通过顺式作用在转录或转录后水平调控基因表达[13-14]。Kim等[15]曾鉴定出547个与禾谷镰孢()子实体形成相关的lncRNA,并发现其中多数lncRNA以顺式作用调控mRNA的表达。LncRNA在酿酒酵母()和裂殖酵母()中可通过顺式作用调节的启动子,从而影响酵母的生殖细胞分化[16]。此外,lncRNA可作为微小RNA(microRNA,miRNA)前体,通过一系列加工和剪切形成成熟的miRNA,从而调控下游靶mRNA的表达[17-18]。再者,含有miRNA反应元件(miRNA response element,MRE)的lncRNA还能作为竞争性内源RNA(competing endogenous RNA,ceRNA)靶向结合miRNA,从而对靶基因的表达进行间接调控[13,19]。Qin等[20]虽然早在2006年组装并公布了球囊菌ARSEF7405菌株的基因组序列,但并未同时公布基因功能注释信息,导致球囊菌的分子生物学和组学研究举步维艰。前人对白垩病的研究主要涉及病原鉴定[21]、流行病学[22]、形态学[23]和快速诊断[24]等方面。2016年,Shang等[25]从头组装和注释了球囊菌ARSEF 7405菌株的参考基因组(AAP 1.0),为深入开展球囊菌的分子生物学和组学研究打下了基础。随着高通量测序技术特别是基于链特异性建库的lncRNA-seq技术的发展,人们已在动物[26]、植物[27]、细菌[28]、真菌[29]、病毒[30]中鉴定到大量lncRNA。【本研究切入点】笔者团队前期已利用lncRNA-seq技术对球囊菌的孢子和菌丝混合样品进行测序,预测和分析了球囊菌的lncRNA和环状RNA(circular RNA,circRNA)的数量、种类及结构特征,并验证了二者的真实表达[31-32]。然而,球囊菌菌丝和孢子lncRNA的数量、结构、表达谱差异还没有明确,lncRNA与菌丝和孢子生长、发育、生殖和致病性的关系仍不清楚。【拟解决的关键问题】利用基于链特异性建库的lncRNA-seq技术对球囊菌的孢子和菌丝分别进行测序,通过生物信息学方法筛选并深入分析孢子和菌丝的共有lncRNA、特有lncRNA和差异表达lncRNA(differentially expressed lncRNA,DElncRNA),以期解析球囊菌孢子和菌丝lncRNA的数量、种类和表达谱差异,挖掘菌丝生长、孢子萌发、杂交产孢、毒力因子等相关的lncRNA并探讨其潜在功能。
1 材料与方法
试验于2019年6月至2020年1月在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。
1.1 供试球囊菌
供试球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离和保存[33-34]。
1.2 球囊菌培养及测序样品制备
参照本课题组前期建立的方法[35],将实验室保存的球囊菌菌株接种到马铃薯葡萄糖琼脂(potato dextrose agar,PDA)培养基,移入恒温生化培养箱33℃条件下培养,培养7 d后分别制备纯化的球囊菌菌丝和孢子。将上述菌丝样品和孢子样品分别命名为AaM和AaS。
1.3 RNA提取、cDNA链特异性文库构建及深度测序
从超低温冰箱中取出菌丝和孢子样品,分别置于两个干净的研钵,加入液氮充分研磨。利用AxyPrepTMMultisource Total RNA Miniprep试剂盒(TaKaRa公司,日本)分别提取球囊菌菌丝和孢子的总RNA。参照Guo等[31]的方法构建链特异性cDNA文库。采用Illumina HiSeqTM4000平台对上述构建好的文库进行双端测序。测序数据已上传到NCBI SRA数据库,BioProject号:PRJNA560452。
1.4 高通量测序数据过滤质控与lncRNA预测
参照GUO等[31]的方法对下机的原始读段(raw reads)进行严格质控以保证测序数据的质量。利用reads比对工具Bowtie2[36]将质控得到的有效读段(clean reads)比对到核糖体数据库(允许错配率为0),去除比对上的clean reads。再利用Tophat2[37]将剩余的clean reads比对到球囊菌的参考基因组(AAP 1.0)上,得到比对上的有效读段(mapped clean reads)。使用Cufflinks软件[38]进行转录本的重构,根据组装出来的转录本在参考基因组上的位置,筛选长度≥200 bp且外显子数目≥2的转录本,作为候选转录本。联用CPC[39]和CNCI[40]软件预测候选转录本的编码能力,同时将转录本对比到SwissPort蛋白数据库(https://www.uniprot.org),将没有编码能力且没有蛋白注释信息的转录本预测为lncRNA,取二者预测结果的交集作为可信的lncRNA。
1.5 LncRNA的Venn分析和差异分析
利用Omicshare在线工具集合(https://www. omicshare.com)对AaM与AaS的lncRNA进行Venn分析,筛选出两组样品的共有和特有lncRNA。使用edgeR软件[41]对AaM和AaS的lncRNA进行差异分析,根据≤0.05(FDR矫正)且|log2fold change (FC)|≥1标准筛选DElncRNA。
1.6 共有lncRNA、特有lncRNA和DElncRNA的顺式作用分析
LncRNA的顺式作用指的是lncRNA的功能与其上下游相邻的蛋白编码基因有关,处于上下游的lncRNA可以与共表达基因的顺式作用元件或者3′UTR存在交集,从而起到对靶基因的调控作用[42]。利用Blast软件将共有lncRNA、特有lncRNA和DElncRNA的上下游基因分别映射到GO和KEGG数据库,对映射上的GO条目数、KEGG通路数及相应的基因数分别进行统计。
1.7 DElncRNA的ceRNA调控网络构建分析及miRNA前体预测
笔者团队前期同时利用small RNA-seq(sRNA- seq)技术对球囊菌纯化菌丝、纯化孢子样品分别进行测序,获得了高质量的miRNA组学数据[43],可为本研究中mRNA、DEmRNA、lncRNA和DElncRNA的靶向预测及ceRNA调控网络构建提供数据支持。sRNA-seq的原始数据已上传NCBI SRA数据库,BioProject号:PRJNA560456。
参照本课题组前期建立的方法[44],利用TargetFind软件[45]预测lncRNA与miRNA、miRNA与mRNA的靶向结合关系,并构建及可视化lncRNA- miRNA-mRNA和DElncRNA-DEmiRNA-DEmRNA调控网络。将预测出的lncRNA对比到miRBase(version 21),寻找潜在的miRNA前体,选取对其中覆盖度>90%的lncRNA,同时利用miRPara(version 6.3)软件预测miRNA及其前体。
1.8 DElncRNA实时荧光定量PCR(RT-qPCR)验证
随机选取4个DElncRNA(TCONS_00006988、TCONS_00003707、TCONS_00001814、TCONS_00007359)进行RT-qPCR验证。参照郭睿等[26]方法设计合成DElncRNA的特异性上游引物和下游引物。选择(5417)作为内参基因。相关的引物信息详见表1。利用RNA提取试剂盒(TaKaRa公司,日本)分别提取球囊菌菌丝和孢子的总RNA,使用cDNA第1链合成试剂盒(上海翊圣,中国)对提取的RNA进行反转录,得到相应的cDNA作为qPCR的模板。qPCR反应体系为20 μL:SYBR Green Dye 10 μL,上下游引物各1 μL(10 μmol·L-1),cDNA模板1 μL,DEPC水7 μL。配制的反应体系在ABI QuanStudio 3荧光定量PCR仪(ABI公司,美国)中进行反应。qPCR程序:94℃预变性5 min;94℃变性30 s,53℃退火40 s,72℃延伸30 s,共40个循环。每组qPCR反应设置3次技术重复。采用2-∆∆Ct方法计算DElncRNA的相对表达量。利用GraphPad Prism 7软件(GraphPad公司,美国)进行数据的Student’s-test和相关绘图。

表1 本研究所用的引物
2 结果
2.1 LncRNA-seq数据的质控与评估
AaM和AaS通过测序得到的raw reads分别为108 614 646和105 675 408条,质控后得到的clean reads分别为107 780 032与104 621 402条;20分别为98.76%和98.72%,30分别达到95.84%和95.78%(表2)。上述结果表明测序数据质量良好,可用于进一步分析。

表2 LncRNA-seq数据概览
2.2 球囊菌菌丝与孢子中lncRNA的数量和种类
基于AaM的测序数据预测出740个lncRNA,其中基因间区lncRNA、双向lncRNA、反义链lncRNA和正义链lncRNA分别为302、199、66和31个;基于AaS的测序数据预测出811个lncRNA,包括324个基因间区lncRNA,230个双向lncRNA,68个反义链lncRNA和36个正义链lncRNA。Venn分析结果显示AaM和AaS包含701个共有lncRNA,特有lncRNA的数量分别为39和110个。
2.3 球囊菌菌丝与孢子共有和特有lncRNA的筛选及上下游基因的数据库注释
GO数据库注释结果显示,701个共有lncRNA可以通过顺式作用潜在调控3 992个上下游基因,这些上下游基因可注释到细胞进程(542)、代谢进程(539)和催化活性(477)等42个GO功能条目(图1-A);AaM的特有lncRNA的上下游基因可注释到细胞进程(49)、代谢进程(41)和催化活性(39)等32个功能条目(图1-B);AaS的特有lncRNA的上下游基因可注释到代谢进程(130)、细胞进程(129)和催化活性(117)等36个功能条目(图1-C)。括号内的数字代表注释到该条目(或通路)的上下游基因数。
KEGG数据库注释结果显示,共有lncRNA的上下游基因还可注释到代谢途径(260)、次生代谢物的生物合成(122)及抗生素的生物合成(92)等117条通路(图2-A);AaM特有lncRNA的上下游基因能注释到代谢途径(21)、次生代谢产物的生物合成(8)及嘌呤代谢(6)等56条通路(图2-B);AaS特有lncRNA的上下游基因能注释到代谢途径(73)、次生代谢产物的生物合成(26)及抗生素的生物合成(20)等95条通路(图2-C)。
2.4 AaM vs AaS比较组中DElncRNA的筛选及功能和通路注释
差异分析结果显示,AaM vs AaS比较组包含255个DElncRNA,其中上调和下调lncRNA的数量分别为181和74个;上调幅度最大的前3位分别是TCONS_00004900(log2fc=13.975)、TCONS_00006709(log2fc=13.785)和TCONS_00001694(log2fc= 13.258);下调幅度最大的前3位分别是TCONS_ 00004890(log2fc=-13.374)、TCONS_00005341(log2fc=-12.481)和TCONS_00008852(log2fc=-12.388)。
255个DElncRNA共预测出1 479个上下游基因,包括773个上游基因和706个下游基因。这些上下游基因可注释到41个功能条目,包括细胞(203)和细胞组件(203)等12个细胞组分相关条目;催化活性(248)和结合(171)等10个分子功能相关条目;代谢进程(275)和细胞进程(270)等19个生物学进程相关条目;此外,还可注释到代谢途径(137)及次生代谢产物的生物合成(61)等107条通路。括号内的数字代表注释到该条目(或通路)的上下游基因数。
2.5 共有lncRNA、特有lncRNA和DElncRNA作为miRNA前体的分析
从菌丝和孢子的22个共有lncRNA中预测出41个miRNA前体序列;从孢子的1个特有lncRNA中预测出5个miRNA前体序列;从菌丝的特有lncRNA中未能预测出miRNA前体序列;从5个DElncRNA中预测出13个miRNA前体序列;进一步分析发现这些miRNA前体序列均含有典型的颈环结构,并且包含成熟miRNA的序列。图3展示了DElncRNA中预测出的5个miRNA前体序列及相应的成熟miRNA序列。
2.6 菌丝lncRNA、孢子lncRNA和DElncRNA的调控网络构建及分析
利用Cytoscape软件对菌丝全部lncRNA、孢子全部lncRNA的调控网络进行可视化,分析结果显示菌丝的10个lncRNA靶向结合8个miRNA,上述miRNA靶向结合77个mRNA(图4-A);孢子的8个lncRNA靶向结合7个miRNA,这些miRNA靶向结合87个mRNA(图4-B);LncRNA和mRNA位于调控网络的外周,而miRNA处于调控网络的核心。
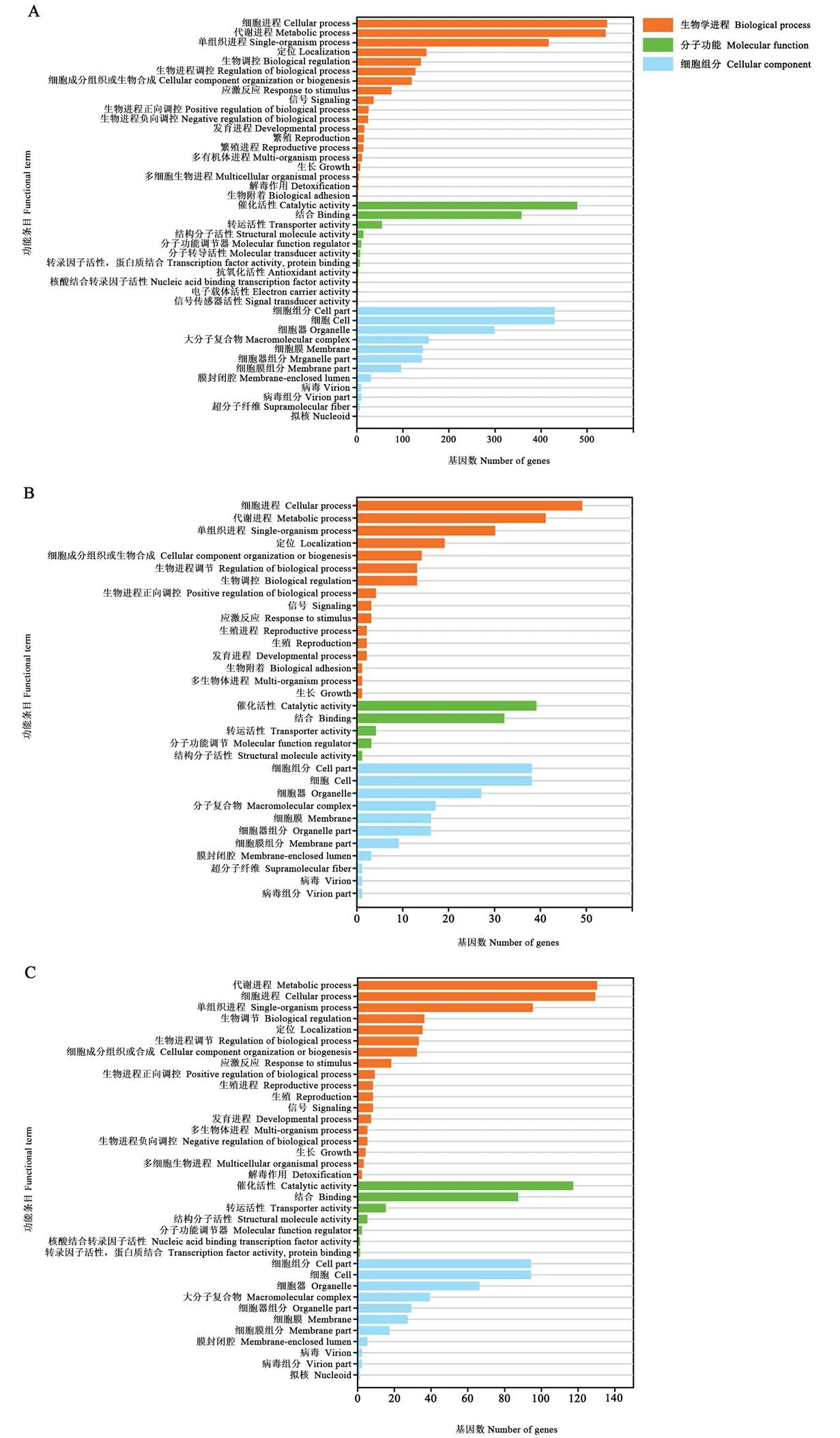
A:AaM与AaS共有lncRNA上下游基因的GO数据库注释GO database annotation of upstream and downstream genes of common lncRNAs in AaM and AaS;B:AaM特有lncRNA的GO数据库注释GO database annotation of upstream and downstream genes of specific lncRNAs in AaM;C:AaS特有lncRNA的GO数据库注释GO database annotation of upstream and downstream genes of specific lncRNAs in AaS
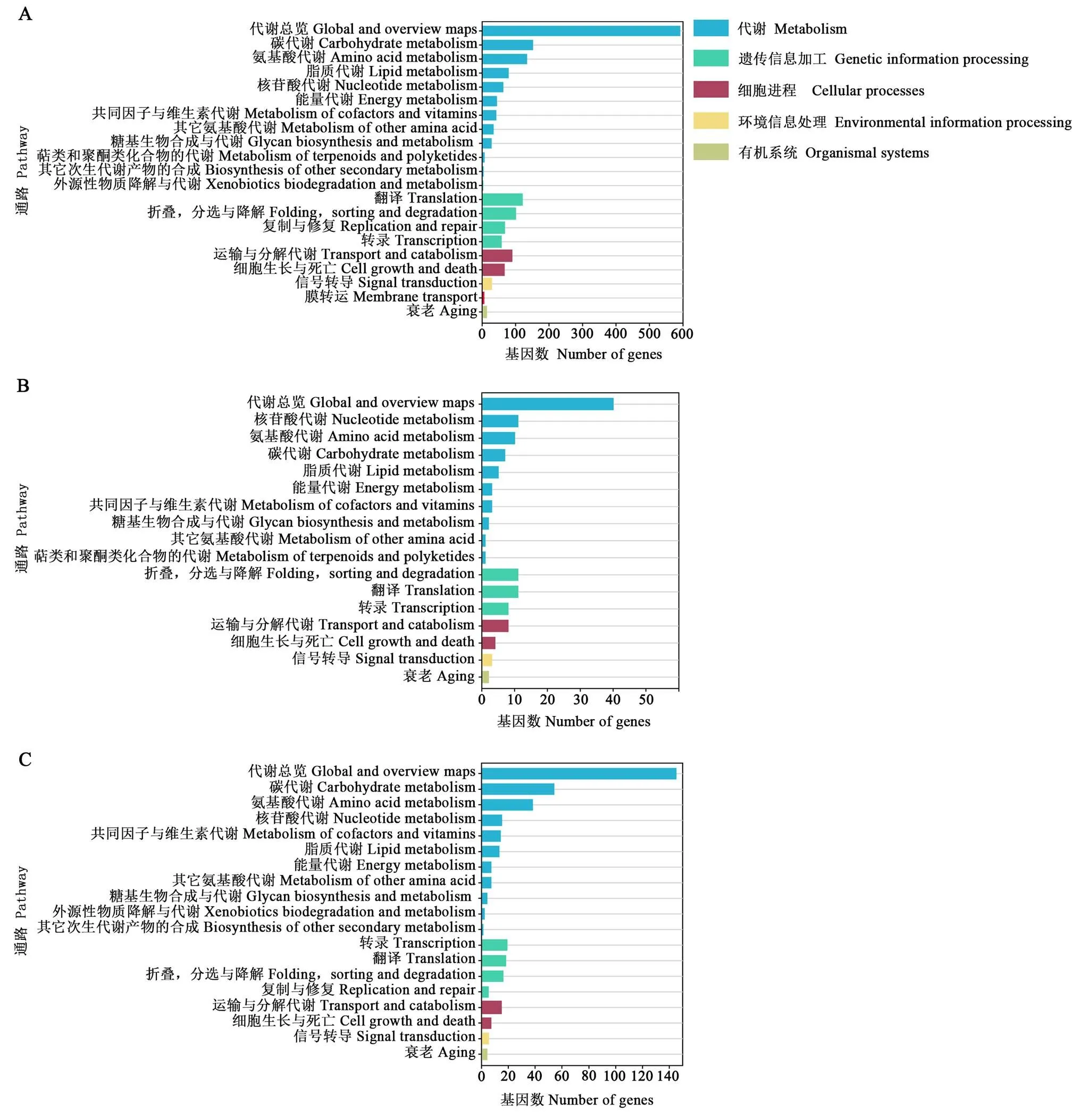
A:AaM与AaS共有lncRNA上下游基因的KEGG富集分析KEGG database annotation of upstream and downstream genes of common lncRNAs in AaM and AaS;B:AaM特有lncRNA的KEGG富集分析KEGG database annotation of upstream and downstream genes of specific lncRNAs in AaM;C:AaS特有lncRNA的KEGG富集分析KEGG database annotation of upstream and downstream genes of specific lncRNAs in AaS
进一步对DElncRNA与DEmiRNA、DEmRNA的调控网络进行构建和分析,结果显示DElncRNA TCONS_00008630与TCONS_00009302共同靶向miR-4968-y,进而调控10个DEmRNA(图4-C)。上述10个DEmRNA可注释到11个功能条目,包括细胞(2)、细胞组分(2)、细胞器(2)、细胞器组分(1)、腔上包膜(1)、结合(3)、催化活性(2)、代谢进程(3)、细胞进程(3)、单细胞进程(1)、生物调节(1);此外还能注释到11条通路,包括代谢途径(2)、次生代谢物的生物合成(1)、抗菌活性(1)、不同环境中的微生物代谢(1)、氨基酸生物合成(1)、嘌呤代谢(1)、嘧啶代谢(1)、丙氨酸、天门冬氨酸和谷氨酸代谢(1)、RNA聚合酶(1)、碱基切除修复(1)、氮代谢(1)。括号内的数字代表注释到该条目(或通路)的上下游基因数。
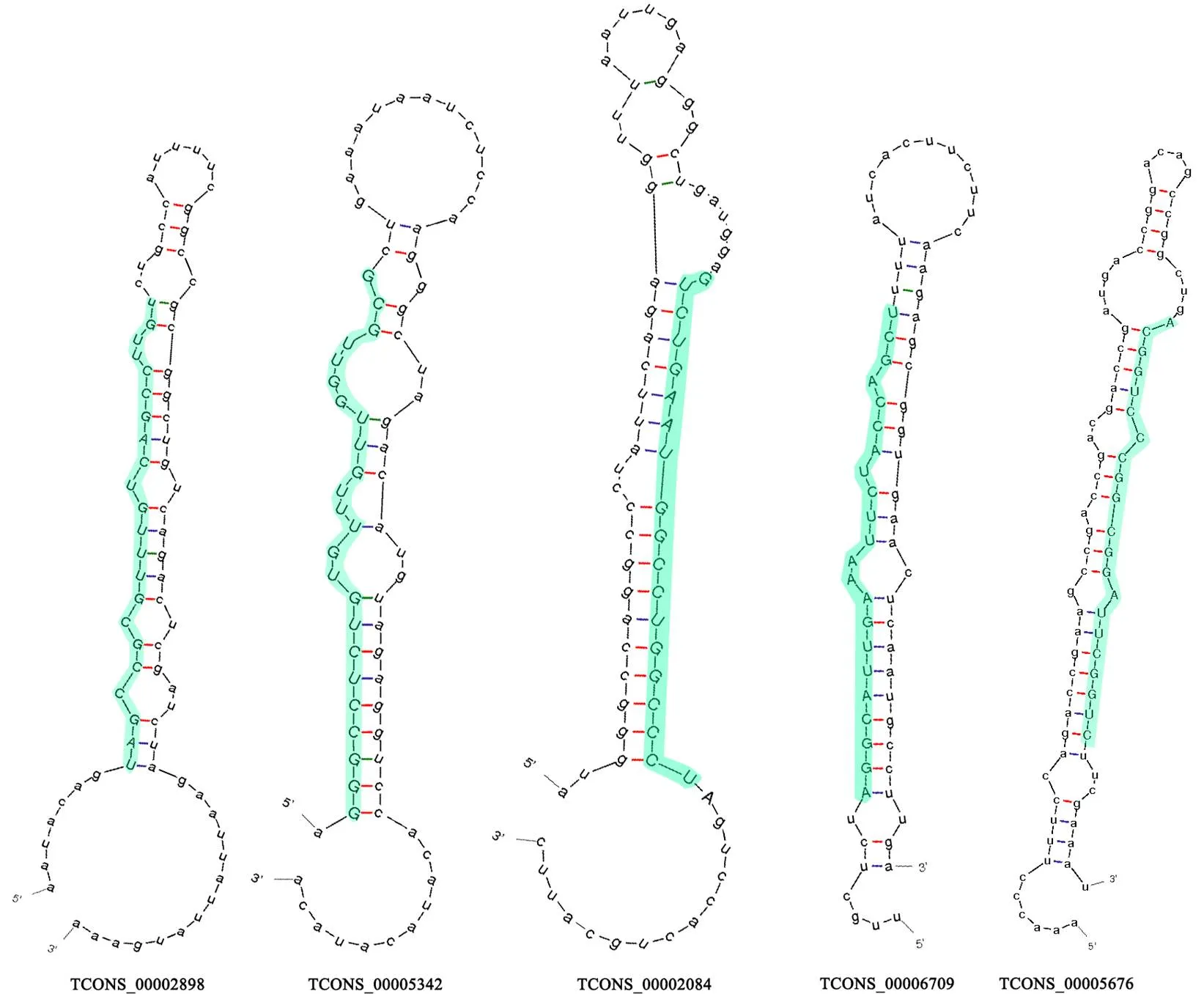
绿色碱基代表成熟miRNA的序列Green bases indicate sequences of mature miRNAs
2.7 DElncRNA 的RT-qPCR验证
随机挑选4个DElncRNA进行RT-qPCR验证,结果表明DElncRNA的变化趋势均与测序结果一致(图5),证明本研究的测序数据和lncRNA的差异表达真实可靠。
3 讨论
近年来,较多的研究结果证实lncRNA在真核生物的生理、代谢、免疫和疾病等过程扮演重要角色[46-47]。本研究首先在实验室条件下对球囊菌进行纯培养,然后分离得到纯化菌丝样品和纯化孢子样品,进而利用基于链特异性建库的lncRNA-seq技术对二者分别进行测序,通过生物信息学方法从菌丝和孢子中分别预测出740和811个lncRNA,其中701个lncRNA为菌丝和孢子所共有,39和110个lncRNA在二者中特异性表达。鉴于lncRNA表达具有物种、组织、发育时期和胁迫阶段特异性[48],上述共有lncRNA可能在球囊菌的不同存在形态中发挥一般性的调控功能,而特有lncRNA在菌丝和孢子中分别发挥不同的调控功能。本研究中,球囊菌菌丝和孢子处于离体环境,推测仍有可能存在一些lncRNA在球囊菌侵染蜜蜂幼虫的过程中特异性表达。菌丝和孢子中数量最多的lncRNA类型均为基因间区lncRNA,而内含子lncRNA的数量很少。这与东方蜜蜂微孢子虫()、罗伯茨绿僵菌()、酿酒酵母[17,49]等真菌的lncRNA主要类型相似。对于菌丝和孢子共有lncRNA的上下游基因,分别有542、539和477个涉及细胞进程、代谢进程和催化活性;74和35个涉及应激反应和信号;14与13个涉及生殖和生殖进程。这表明共有lncRNA可能通过顺式作用调控上下游基因的表达,从而参与球囊菌菌丝和孢子的细胞生命活动、新陈代谢、信号转导和有性生殖等生物学过程。此外,共有lncRNA的上下游基因还能注释到117条通路,其中有133个上下游基因注释到酪氨酸代谢(9)等14条氨基酸代谢相关通路;151个注释到糖酵解/糖异生通路(24)等15条碳水化合物代谢相关通路;78个注释到甘油磷脂代谢(17)等13条脂质代谢相关通路;62个注释到嘌呤代谢(34)和嘧啶代谢(28);还有43个上下游基因可注释到4条能量代谢相关通路,包括氧化磷酸化(22)、甲烷代谢(10)、硫代谢(7)及氮代谢(4)。上述结果再次说明共有lncRNA通过顺式作用广泛参与球囊菌菌丝和孢子的物质代谢和能量代谢方面的调控。

图4 菌丝lncRNA(A)、孢子lncRNA(B)和DElncRNA(C)的调控网络
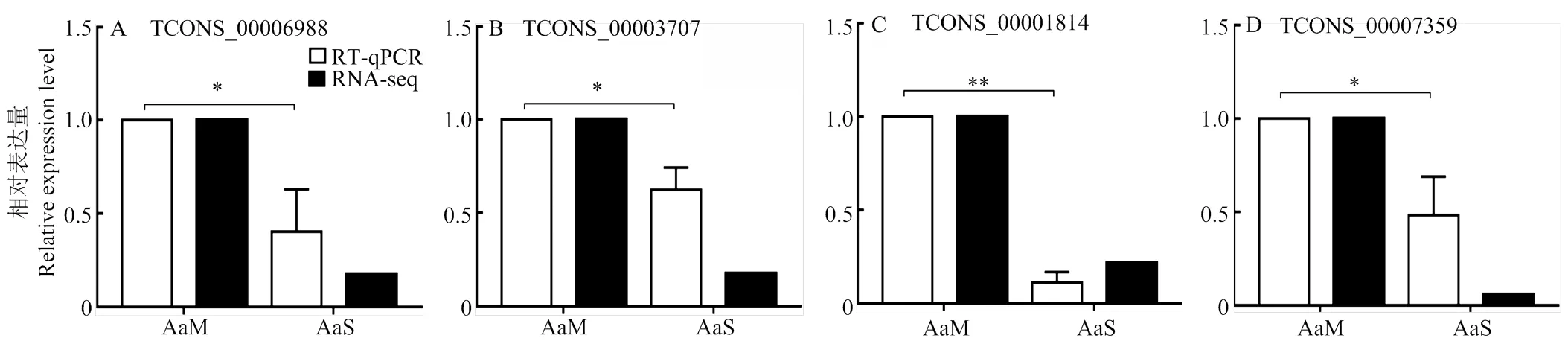
*: p<0.05; **: p<0.01
3.1 球囊菌菌丝和孢子的共有lncRNA与自噬通路具有潜在的调控关系
自噬是真核生物非常重要且高度保守的蛋白质降解过程,通过自噬体将细胞中的细胞器、蛋白质及其他生物大分子包裹后运送到具有降解作用的细胞器进行降解并重新利用,从而使得细胞在营养缺乏的调节下维持基本生存[50]。自噬已被证明与真菌的孢子产生存在直接关联[51]。稻瘟病菌()是严重危害水稻的真菌病原,Liu等[52]在稻瘟病菌中鉴定到与自噬相关的基因,并发现该基因与稻瘟病菌的分生孢子形成直接相关;Deng等[53]发现缺失的稻瘟病菌菌株产孢能力显著降低。球囊菌通过异宗配合进行有性生殖,PDA培养基上生长早期的球囊菌菌丝为白色,当异宗菌丝接触发生配合后才会形成内含大量孢子的黑色孢子囊。本研究中,30个共有lncRNA(TCONS_00008713、TCONS_00008558和TCONS_00003019等)的25个上下游基因(KZZ86589.1、KZZ86859.1和KZZ86861.1等)可注释到自噬通路,说明这些共有lncRNA可能通过顺式作用对自噬相关的上下游基因进行表达调控,从而影响球囊菌的杂交产孢。
3.2 球囊菌菌丝特有lncRNA可能参与病原对蜜蜂幼虫的侵染过程
昆虫中肠内侧有一层致密的围食膜,主要由几丁质、多糖和蛋白质等成分组成,能够将上皮细胞与食物隔开,构成昆虫抵御病原微生物入侵的一道物理屏障[54]。在被感染的蜜蜂幼虫肠道内,球囊菌菌丝大量生长的过程一方面通过机械作用刺穿围食膜和肠壁,同时合成和分泌一些几丁质酶、水解酶、脂酶和蛋白酶等毒力因子共同作用分解宿主中肠围食膜,协同促进病原的增殖和侵染[3]。泛素是一类存在于多数真核细胞中的高度保守的蛋白质,对于蛋白合成与降解至关重要[55]。本研究中,菌丝的特有lncRNA的2个上下游基因(KZZ91868.1和KZZ97813.1)可注释到泛素介导的蛋白水解通路,另有1个上下游基因(KZZ95233.1)可注释到蛋白酶体通路,表明调控KZZ91868.1、KZZ97813.1和KZZ95233.1的菌丝特有lncRNA(TCONS_00004660、TCONS_00000056和TCONS_00002376)参与调节病原侵染宿主过程对围食膜的分解。真菌侵染昆虫的过程中会通过产生一些次生代谢产物(例如毒素)促进自身侵染。卵孢白僵菌()[56]在侵染血黑蝗()过程中分泌的草酸能够溶解宿主体壁;金龟子绿僵菌()[57]在侵染大蜡螟()过程中产生的细胞松弛素可降低血淋巴的吞噬能力,有助于其自身在宿主体内的存活。本研究中,4个菌丝特有lncRNA调控的8个上下游基因可注释到次生代谢产物的生物合成,包括KZZ86645.1(核糖磷酸焦磷酸激酶1编码基因)和KZZ87421.1(2-异丙基苹果酸合成酶编码基因)等,说明这些特有lncRNA可能参与球囊菌侵染过程毒力因子的合成与分泌。但需要强调的是,本研究测序材料来源于实验室条件下获得的球囊菌纯培养,其表达的lncRNA必然与处于侵染过程的球囊菌所表达的lncRNA存在差异,因此上述结果显示出的是部分特有lncRNA与球囊菌致病力的潜在关联,若要明确二者的直接关系,需要通过对处于侵染过程的球囊菌lncRNA组学数据、纯化孢子的lncRNA组学数据进行比较分析,从而筛选和挖掘DElncRNA的相关信息。目前,笔者团队已利用基于链特异性建库的lncRNA-seq技术对正常及球囊菌胁迫的意蜂幼虫肠道、中蜂幼虫肠道进行深度测序,获得了高质量的lncRNA组学数据(未发表数据),下一步将通过比对宿主参考基因组过滤掉来源于宿主的lncRNA组学数据,再将剩余数据比对球囊菌参考基因组,从而获得来源于球囊菌本身的lncRNA组学数据,进而结合本研究中球囊菌纯化孢子的lncRNA组学数据进行细致深入的比较分析。
3.3 球囊菌在孢子状态仍具有lncRNA转录及较低水平的代谢活动
真菌孢子是一种休眠态的存在形式,相对于营养状态的菌丝,人们对于孢子中核酸的转录、翻译和代谢方面的认识较为有限。球囊菌的孢子对不良环境有很强的抵抗力,可以存活并保持感染力超过15年以上[58]。本研究发现,仅在孢子中特异性表达的110个lncRNA可通过顺式作用调控672个上下游基因,其中有300个上下游基因可注释到物质和能量代谢相关通路,包括络氨酸代谢(6)等12条氨基酸代谢通路,氨基酸糖和核苷酸糖代谢(7)等15条碳代谢通路,氧化磷酸化(4)等4条能量代谢通路,以及脂肪酸降解(2)等9条脂质代谢通路。上述结果暗示球囊菌在其孢子状态仍进行着较低水平的物质和能量代谢活动,部分lncRNA参与了上述代谢活动的调控。东方蜜蜂微孢子虫是另一种广泛感染世界各地蜂群的蜜蜂真菌病原。笔者团队前期研究发现东方蜜蜂微孢子虫的孢子中同样存在一定水平的转录活动[29]。这与本研究的结果相似,表明蜜蜂真菌病原的孢子内部同样存在低水平的转录及新陈代谢活动,推测这对于维持孢子的完整性和活力是必要的。
3.4 球囊菌菌丝和孢子的DElncRNA具有通过调控MAPK信号通路影响病原致病性的潜在功能
本研究中,共鉴定到181个上调lncRNA和74个下调lncRNA。这些DElncRNA的上下游基因可注释到41个功能条目,其中分别有3、10、21和36个上下游基因注释到生长、发育进程、生殖和应激反应,说明DElncRNA参与上述生物学过程的调控。真菌通过信号通路感受外界环境变化进而调整其生理状态[59];此外,信号通路与真菌的繁殖、生长和毒力密切相关[60]。真菌中,MAPK信号通路是一条具有较高保守性的关键信号通路,已被证实在酿酒酵母的交配、侵染、增殖、细胞壁完整性、渗透调节和孢子形成等方面起到重要作用[61]。新月弯孢()是一种侵染玉米的真菌,Ni等[62]通过对该病原的2个MAPK信号通路基因和进行研究,发现与病原孢子形成和致病性有关,而与细胞壁形成及病原侵染过程相关。中华蜜蜂()和意大利蜜蜂()是我国养蜂生产中使用的主要蜂种,前者对球囊菌抗性较强,后者则较为易感。笔者团队前期研究发现,对于侵染意蜂幼虫的球囊菌,有48个基因富集在MAPK通路且呈上调表达趋势,而侵染中蜂的球囊菌只有11个下调基因富集于MAPK通路[13]。本研究发现,11个DElncRNA(TCONS_00000156、TCONS_00006998和TCONS_00008039等)通过顺式作用调控的9个上下游基因注释到MAPK信号通路,暗示这些DElncRNA可能在球囊菌侵染蜜蜂幼虫的过程中参与对MAPK信号通路的调控,从而影响病原的致病性。
3.5 球囊菌菌丝和孢子的DElncRNA可能作为ceRNA调控物质和能量代谢以及遗传信息传递
Salmena等[63]提出了ceRNA假说,该假说认为具有miRNA反应元件的转录本,例如lncRNA、circRNA、假基因等均可以通过竞争结合miRNA减弱miRNA对mRNA的抑制或降解作用。此后,该假说已被较多的研究[19,64]所证实。本研究中构建的DElncRNA-DEmiRNA-DEmRNA调控网络并不复杂,其中仅有2个DElncRNA TCONS_00008630(log2fc= -1.67)和TCONS_00009302(log2fc=-11.48)靶向结合miR-4968-y(log2fc=3.37),进而调控10个靶mRNA,这些靶mRNA涉及能量代谢(1)、复制与修复(1)、转录(1)、氨基酸代谢(1)等活动。上述结果表明球囊菌菌丝和孢子的DElncRNA充当ceRNA调控球囊菌的物质和能量代谢以及遗传信息传递。
4 结论
通过对球囊菌菌丝和孢子共有lncRNA、特有lncRNA和DElncRNA的全面分析和深入探讨,解析了菌丝和孢子中lncRNA的数量、种类和表达谱差异,并揭示了部分lncRNA可能通过顺式作用和ceRNA机制参与调控球囊菌菌丝和孢子的生长、发育和生殖等生物学过程。研究结果为深入理解球囊菌的基础生物学提供了重要的参考信息,也为深入研究lncRNA及其调控网络介导球囊菌对蜜蜂幼虫的侵染过程提供了必要的数据基础。
[1] Calderón RA, Rivera G, Sánchez LA, Zamora LG. Chalkbrood () and some other fungi associated with Africanized honey bees () in Costa Rica. Journal of Apicultural Research, 2004, 43(4): 187-188.
[2] WOOD M. Microbes help bees battle chalkbrood. Agricultural Research, 1998, 46(8): 16-17.
[3] ARONSTEINK K A, MURRAY D. Chalkbrood disease in honey bees.Journal of Invertebrate Pathology, 2010, 103(Suppl.1): 20-29.
[4] KAPRANOV P, CHENG J, DIKE S, NIX D A, Duttagupta R, Willingham A T, Stadler P F, HERTEL J, HACKERMÜLLER J, HOFACKER I L,. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science, 2007, 316(5830): 1484-1488.
[5] PONTIER D B, GRIBNAU J. Xist regulation and function explored. Human Genetics, 2011, 130(2): 223-236.
[6] KINO T, HURT D E, ICHIJO T, NADER N, CHROUSOS G P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Science Signaling, 2010, 3(107): ra8.
[7] HEO J B, SUNG S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA.Science, 2011, 331(6013): 76-79.
[8] TSAI M C, MANOR O, WAN Y, MOSAMMAPARAST N, WANG J K, LAN F, SHI Y, SEGAL E, CHANG H Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science, 2010, 329(5992): 689-693.
[9] MERCER T R, MATTICK J S. Structure and function of long noncoding RNAs in epigenetic regulation.Nature Structural and Molecular Biology, 2013, 20(3): 300-307.
[10] LI M, SUN X, CAI H, SUN Y, PLATH M, LI C, LAN X, LEI C, LIN F, BAI Y, CHEN H. Long non-coding RNA adncr suppresses adipogenic differentiation by targeting miR-204. Biochimica et Biophysica Acta, 2016, 1859(7): 871-882.
[11] ST LAURENT G, WAHLESTEDT C, KAPRANOV P. The landscape of long noncoding RNA classification. Trends in Genetics, 2015, 31(5): 239-251.
[12] LI Z X, HAN K W, ZHANG D F, CHEN J G, XU Z, HOU L J. The role of long noncoding RNA in traumatic brain injury. Neuropsychiatric Disease and Treatment, 2019, 15: 1671-1677.
[13] 郭睿, 王海朋, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 赵红霞, 陈大福. 蜜蜂球囊菌的microRNA鉴定及其调控网络分析. 微生物学报, 2018, 58(6): 1077-1089.
GUO R, WANG H P, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, ZHAO H X, CHEN D F. Identification ofmicroRNAs and investigation of their regulation networks. Acta Microbiologica Sinica, 2018, 58(6): 1077-1089. (in chinese)
[14] KOPP F, MENDELL J T. Functional classification and experimental dissection of long noncoding RNAs. Cell, 2018, 172(3): 393-407.
[15] KIM W, Miguel-Rojas C, Wang J, TownsendJ P, Trail F. Developmental dynamics of long noncoding RNA expression during sexual fruiting body formation in. mbio, 2018, 9(4): e01292-18.
[16] HiriartE, VerdelA. Long noncoding RNA-based chromatin control of germ cell differentiation: a yeast perspective. Chromosome Research, 2013, 21(6/7): 653-663.
[17] CHEN D F, CHEN H Z, DU Y, ZHOU D D, GENG S H, WANG H P, WAN J Q, XIONG C L, ZHENG Y Z, GUO R. Genome-wide identification of long non-coding RNAs and their regulatory networks involved inresponse toinfection. Insects, 2019, 10(8): 245.
[18] WILUSZ J E, SUNWOO H, SPECTOR D L. Long noncoding RNAs: functional surprises from the RNA world. Genes and Development, 2009, 23(13): 1494-1504.
[19] FAN G Q, WANG Z, ZHAI X Q, CAO Y B. ceRNA cross-talk in paulownia witches’ broom disease. International Journal of Molecular Sciences, 2018, 19(8): 2463.
[20] QIN X, EVANS J D, ARONSTEIN K A, MURRAY K D, WEINSTOCK G M. Genome sequences of the honey bee pathogensand. Insect Molecular Biology, 2006, 15(5): 715-718.
[21] SPILTOIR C F. Life cycle of(). American Journal of Botany, 1955, 42(6): 501-508.
[22] FLORES J M, SPIVAK M, GUTIÉRREZ I. Spores ofcontained in wax foundation can infect honeybee brood. Veterinary Microbiology, 2005, 108(1/2): 141-144.
[23] SKOU J P. More details in support of the classAscosphaeromycetes. Mycotaxon, 1988, 31(1): 191-198.
[24] Anderson D, Giacon H Search articles by 'Giacon H' Giacon H, Gibson N Search articles by 'Gibson N' Gibson N. Detection and thermal destruction of the chalkbrood fungus () in honey. Journal of Apicultural Research, 1997, 36(3/4): 163-168.
[25] SHANG Y F, XIAO G H, ZHENG P, CEN K, ZHAN S, WANG C S. Divergent and convergent evolution of fungal pathogenicity. Genome Biology and Evolution, 2016, 8(5): 1374-1387.
[26] 郭睿, 耿四海, 熊翠玲, 郑燕珍, 付中民, 王海朋, 杜宇, 童新宇, 赵红霞, 陈大福. 意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析. 中国农业科学, 2018, 51(18): 3600-3613.
GUO R, GENG S H, XIONG C L, ZHENG Y Z, FU Z M, WANG H P, DU Y, TONG X Y, ZHAO H X, CHEN D F. Differential expression analysis of long non-coding RNAs during the developmental process ofworker’s midgut. Scientia Agricultura Sinica, 2018, 51(18): 3600-3613. (in chinese)
[27] Flórez-ZapataN M V, Reyes-ValdésM H, MartínezO. Long non-coding RNAs are major contributors to transcriptome changes in sunflower meiocytes with different recombination rates. BMC Genomics, 2016, 17: 490.
[28] YANG L, LONG Y P, LI C, CAO L, GAN H Y, HUANG K L, JIA Y J. Genome-wide analysis of long noncoding RNA profile in human gastric epithelial cell response to. Japanese Journal of Infectious Diseases, 2015, 68(1): 63-66.
[29] GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, LIANG Q, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, KUMAR D. First identification of long non-coding RNAs in fungal parasite. Apidologie, 2018, 49(5): 660-670.
[30] WANG Z Q, ZHAO Y W, ZHANG Y. Viral lncRNA: A regulatory molecule for controlling virus life cycle. Noncoding RNA Research, 2017, 2(1): 38-44.
[31] GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, LI W D, DHIRAJ K, LIANG Q. Identification of long non-coding RNAs in the chalkbrood disease pathogen. Journal of Invertebrate Pathology, 2018, 156: 1-5.
[32] GUO R, CHEN D F, CHEN H Z, FU Z M, XIONG C L, HOU C S, ZHENG Y Z, GUO Y, WANG H P, DU Y, DIAO Q Y. Systematic investigation of circular RNAs in, a fungal pathogen of honeybee larvae. Gene, 2018, 678: 17-22.
[33] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 黄枳腱, 张曌楠, 张璐, 李汶东, 童新宇, 席伟军. 胁迫意大利蜜蜂幼虫肠道的球囊菌的转录组分析. 昆虫学报, 2017, 60(4): 401-411.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, HUANG Z J, ZHANG Z N, ZHANG L, LI W D, TONG X Y, XI W J. Transcriptomic analysis ofstressing larval gut of(Hyemenoptera: Apidae). Acta Entomologica Sinica, 2017, 60(4): 401-411. (in chinese)
[34] 张曌楠, 熊翠玲, 徐细建, 黄枳腱, 郑燕珍, 骆群, 刘敏, 李汶东, 童新宇, 张琦, 梁勤, 郭睿, 陈大福. 蜜蜂球囊菌的参考转录组组装及SSR分子标记开发. 昆虫学报, 2017, 60(1): 34-44.
ZHANG Z N, XIONG C L, XU X J, HUANG Z J, ZHENG Y Z, LUO Q, LIU M, LI W D, TONG X Y, ZHANG Q, LIANG Q, GUO R, CHEN D F.assembly of a reference transcriptome and development of SSR markers for. Acta Entomologica Sinica, 2017, 60(1): 34-44. (in chinese)
[35] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 张曌楠, 黄枳腱, 张璐, 王鸿权, 解彦玲, 童新宇.中华蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学. 中国农业科学, 2017, 50(13): 2614-2623.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, ZHANG Z N, HUANG Z J, ZHANG L, WANG H Q, XIE Y N, TONG X Y. Transcriptome oflarval gut under the stress of. Scientia Agricultura Sinica, 2017, 50(13): 2614-2623. (in chinese)
[36] LANGMEAD B, TRAPNELL C, POP M, SALZBERG S L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 2009, 10(3): R25.
[37] KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R, SALZBERG S L. Tophat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 2013, 14(4): R36.
[38] TRAPNELL C, ROBERTS A, GOFF L, PERTEA G, KIM D, KELLEY D R, PIMENTEL H, SALZBERG S L, RINN J L, PACHTER L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 2012, 7(3): 562-578.
[39] KONG L, ZHANG Y, YE Z Q, LIU X Q, ZHAO S Q, WEI L P, GAO G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research, 2007, 35: W345-349.
[40] SUN L, LUO H T, BU D C, ZHAO G G, YU K T, ZHANG C H, LIU Y N, CHEN R S, ZHAO Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Research, 2013, 41(17): e166.
[41] ROBINSON M D, MCCARTHY D J, SMYTH G K. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 2010, 26(1): 139-140.
[42] SHAO J, CHEN H, YANG D, JIANG M, ZHANG H, WU B, LI J, YUAN L, LIU C. Genome-wide identification and characterization of natural antisense transcripts by strand-specific RNA sequencing in. Scientific Reports, 2017, 7(1): 5711.
[43] 陈华枝, 祝智威, 蒋海宾, 王杰, 范元婵, 范小雪, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 蜜蜂球囊菌菌丝和孢子中微小RNA及其靶mRNA的比较分析. 中国农业科学, 2020, 53(17): 3606-3619.
CHEN H Z, ZHU Z W, JIANG H B, WANG J, FAN Y C, FAN X X, WAN J Q, LU J X, XIONG C L, ZHENG Y Z, FU Z M, CHEN D F, GUO R. Comparative analysis of microRNAs and corresponding target mRNAs inmycelium and spore. Scientia Agricultura Sinica, 2020, 53(17): 3606-3619. (in Chinese)
[44] 熊翠玲, 陈华枝, 陈大福, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 郭睿. 意大利蜜蜂工蜂中肠的环状RNA及其调控网络分析. 昆虫学报, 2018, 61(12): 1363-1375.
XIONG C L, CHEN H Z, CHEN D F, ZHENG Y Z, FU Z M, XU G J, DU Y, WANG H P, GENG S H, ZHOU D D, LIU S Y, GUO R. Analysis of circular RNAs and their regulatory networks in the midgut ofworkers. Acta Entomologica Sinica, 2018, 61(12): 1363-1375. (in chinese)
[45] ALLEN E, XIE Z, GUSTAFSON A M, CARRINGTON J C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants.Cell, 2005, 121(2): 207-221.
[46] ATIANAND M K, FITZGERALD K A. Long non-coding RNAs and control of gene expression in the immune system. Trends in Molecular Medicine, 2014, 20(11): 623-631.
[47] QURESHI I A, MEHLER M F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nature Reviews. Neuroscience, 2012, 13(8): 528-541.
[48] Pauli A, Valen E, Lin M F, Garber M, Vastenhouw N L, Levin J Z, Fan L, Sandelin A, Rinn J L, Regev A, Schier A F. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Research, 2012, 22(3): 577-591.
[49] WANG Z, JIANG Y, WU H, XIE X, HUANG B. Genome-wide identification and functional prediction of long non-coding RNAs involved in the heat stress response in. Frontiers in Microbiology, 2019, 10: 2336.
[50] YORIMITSU T, KLIONSKY D J. Autophagy: molecular machinery for self-eating. Cell Death and Differentiation, 2005, 12(Suppl. 2): 1542-1552.
[51] Fan C L, Chang A N, Liu T B. Role of autophagy in the reproduction of pathogenic fungi. Acta Microbiologica Sinica, 2019, 59(2): 224-234.
[52] LIU X H, ZHAO Y H, ZHU X M, ZENG X Q, HUANG L Y, DONG B, SU Z Z, WANG Y, LU J P, LIN F C. Autophagy-related proteinmoatg14 is involved in differentiation, development and pathogenicity in the rice blast fungus. Scientific Reports, 2017, 7: 40018.
[53] DENG Y Z, RAMOS-PAMPLONA M, NAQVI N I. Autophagy- ssisted glycogen catabolism regulates asexual differentiation in. Autophagy, 2009, 5(1): 33-43.
[54] WANG P, GRANADOS R R. Observations on the presence of the peritrophic membrane in larvaland its role in limiting baculovirus infection. Journal of Invertebrate Pathology, 1998, 72(1): 57-62.
[55] SURESH B, LEE J, KIM K S, RAMAKRISHNA S. The importance of ubiquitination and deubiquitination in cellular reprogramming. Stem Cells International, 2016, 2016:6705927.
[56] Bidochka M J, Khachatourians G G. The implication of metabolic acids produced byin pathogenesis of the migratory grasshopper,. Journal of Invertebrate Pathology, 1991, 58(1): 106-117.
[57] GoTZ P, MATHA V, VILCINSKAS A. Effects of the entomopathogenic fungusand its secondary metabolites on morphology and cytoskeleton of plasmatocytes isolated from the greater wax moth,. Journal of Invertebrate Pathology, 1997, 43(12): 1149-1159.
[58] Jensen A B, Aronstein K, Flores J M, Vojvodic S, Palacio M A, Spivak M. Standard methods for fungal brood disease research. Journal of Apicultural Research, 2013, 52(1): 10.3896/IBRA.1.52.1.13.
[59] Brown A J P, Budge S, Kaloriti D, Tillmann A, Jacobsen M D, Yin Z, Ene I V, Bohovych I, Sandai D, Kastora S, Potrykus J, Ballou E R, Childers D S, Shahana S, Leach M D. Stress adaptation in a pathogenic fungus. Journal of Experimental Biology, 2014, 217(1): 144-155.
[60] So K K, Kim D H. Role of MAPK signaling pathways in regulating the hydrophobin cryparin in the chestnut blight fungusMycobiology, 2017, 45(4): 362-369.
[61] JIANG C, ZHANG X, LIU H Q, XU J R. Mitogen-activated protein kinase signaling in plant pathogenic fungi. PLoS Pathogens, 2018, 14(3): e1006875.
[62] NI X, GAO J X, YU C J, WANG M, SUN J N, LI Y Q, CHEN J. MAPKs and acetyl-CoA are associated withpathogenicity and toxin production in maize.Journal of Integrative Agriculture, 2018, 17(1): 139-148.
[63] SALMENA L, POLISENO L, TAY Y, KATS L, PANDOLFI P P. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell, 2011, 146(3): 353-358.
[64] HUANG M J, ZHAO J Y, XU J J, LI Y, ZHUANG Y F, ZHANG X L. LncRNA ADAMTS9-AS2 controls human mesenchymal stem cell chondrogenic differentiation and functions as a ceRNA. Molecular Therapy-Nucleic Acids, 2019, 18: 533-545.
Comparison and potential functional analysis of long non-coding RNAs betweenmycelium and spore
Chen HuaZhi1, Wang Jie1, Zhu ZhiWei1, Jiang HaiBin1, Fan YuanChan1, Fan XiaoXue1, Wan JieQi1, Lu JiaXuan1, Zheng YanZhen1, Fu ZhongMin1,2,3, Xu GuoJun1, Chen DaFu1,2,3, Guo Rui1,2,3
1College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002;2Apitherapy Research Institution, Fujian Agriculture and Forestry University, Fuzhou 350002;3Engineering Research Center of Processing and Application of Bee Products of Ministry of Education, Fujian Agriculture and Forestry University, Fuzhou 350002
【】is a fungal pathogen that exclusively infects honeybee larvae, causing a sharp decrease of the population of adult honeybees and colony population. Long non-coding RNA (lncRNA), a recently discovered non-coding RNA (ncRNA), plays a vital biological role in various activities such as epigenetics, cell cycle and dose compensation.【】This study aimed to clarify the differences of number, type and expression profile of lncRNAs betweenmycelium and spore, and investigate the potential role of the common lncRNAs, specific lncRNAs and differentially expressed lncRNAs (DElncRNAs). 【】The purified mycelia (AaM) and purified spores (AaS) ofwere respectively sequenced using strand specific library-based lncRNA-seq technology. The expression levels of lncRNAs in AaM and AaS were calculated using FPKM (Fragment Per Kilobase of per Million mapped reads) method. Common lncRNAs and specific lncRNAs were filtered out following Venn analysis. DElncRNAs within AaM vs AaS comparison group were screened out following the standard of≤0.05 and |log2fold change|≥1. Upstream and downstream genes of common lncRNAs, specific lncRNAs and DElncRNAs were aligned against GO and KEGG databases to obtain function and pathway annotations. The competing endogenous RNA (ceRNA) regulation networks of common lncRNAs, specific lncRNAs and DElncRNAs were constructed following target binding relationships, followed by visualization using Cytoscape software. RT-qPCR was performed to verify the reliability of the sequencing data.【】In total, 108 614 646 and 105 675 408 raw reads were gained from AaM and AaS, and after strict filtering, 107 780 032 and 104 621 402 clean reads were obtained, respectively, with20 of 98.76% and 98.72%, and30 of 95.84% and 95.78%. A total of 850 lncRNAs were identified. Seven hundred and one lncRNAs were shared by AaM and AaS, and there were 39 and 110 specific lncRNAs. Viafunction, these shared lncRNAs could regulate 3 992 upstream and downstream genes involving in 42 functional terms such as cellular process, metabolism process and catalytic activity; and 117 pathways such as metabolism pathway, biosynthesis of secondary metabolites and biosynthesis of antibiotics. Specific lncRNAs for AaM and AaS could respectively regulate 243 and 672 upstream and downstream genes. InAaM vs AaS comparison group, 255 DElncRNAs were identified and found to regulate 1 479 upstream and downstream genes, which were associated with 41 functional terms including cellular process, metabolism process and catalytic activity; and 107 pathways including metabolism pathway, biosynthesis of secondary metabolites and biosynthesis of antibiotics. Forty one, five and 13 miRNA precursors were predicted from common lncRNAs, specific lncRNAs and DElncRNAs. The result of regulatory network analysis showed the formation of ceRNA networks of mycelium lncRNAs and spore lncRNAs; ten lncRNAs in mycelium could bind to eight miRNAs, further targeting 77 mRNAs; while eight lncRNAs in spore could link to seven miRNAs, further targeting 87 mRNAs; two DElncRNAs including TCONS_00008630 and TCONS_00009302 could simultaneously target miR-4968-y, further regulating ten mRNAs. The result of RT-qPCR suggested the differential expression trend of four DElncRNAs were in accordance of that in sequencing result, indicating the reliability of our sequencing data.【】Common lncRNAs, specific lncRNAs and DElncRNAs are likely to affect material and energy metabolisms, autophagy, transcription, MAPK signaling pathway, ubiquitin-mediated proteolysis, proteasome and biosynthesis of secondary metabolites, by regulating the expression of upstream and downstream genes, serving as miRNA precursors or ceRNAs, thus regulating the growth, development, reproduction and pathogenicity of
; long non-coding RNA (lncRNA); mycelium; spore; competing endogenous RNA (ceRNA)

10.3864/j.issn.0578-1752.2021.02.018
2020-02-10;
2020-02-25
国家自然科学基金(31702190)、国家现代农业产业技术体系建设专项资金(CARS-44-KXJ7)、福建省自然科学基金(2018J05042)、福建农林大学硕士生导师团队(郭睿)、福建农林大学科技创新专项基金(CXZX2017342,CXZX2017343)、福建省大学生创新创业项目(202010389016,202010389162)
陈华枝,E-mail:CHZ0720@outlook.com。王杰,E-mail:wanglegejie@163.com。陈华枝和王杰为同等贡献作者。通信作者郭睿,E-mail: ruiguo@fafu.edu.cn
(责任编辑 岳梅)
猜你喜欢
动物医学进展(2022年6期)2022-11-26
四川蚕业(2022年1期)2022-06-06
当代水产(2022年1期)2022-04-26
昆明医科大学学报(2021年12期)2021-12-30
云南医药(2021年3期)2021-07-21
现代仪器与医疗(2021年2期)2021-07-21
心肺血管病杂志(2020年5期)2021-01-14
中国瓜菜(2019年8期)2019-09-19
江苏农业科学(2016年11期)2017-03-21
江苏农业科学(2017年1期)2017-02-27
