
PCR 结合斑点杂交技术检测禽多瘤病毒方法的建立及应用
2021-03-07 08:49张富友于晓慧蒋文明孙淑红刘华雷
中国动物检疫 2021年3期
张富友,于晓慧,蒋文明,孙淑红,赵 鹏,刘华雷,李 阳
(1.山东农业大学,山东泰安 271018;2.中国动物卫生与流行病学中心,山东青岛 266114)
鹦鹉幼雏病(budgerigar fledgling disease,BFD)是由禽类多瘤病毒(avian polyomavirus,APV),早期称为鹦鹉幼雏病病毒(budgerigar fledgling disease virus,BFDV)引起的多种鹦鹉幼雏死亡的急性病毒性传染病,主要感染出壳1~3 周的鹦鹉,表现为厌食、消瘦、皮下出血、腹泻和呼吸困难等症状,死亡率可高达80%,并可感染其他多种禽类[1-2]。日龄较小的虎皮鹦鹉(budgerigar,Melopsittacus undulatus)感染APV 时常被称为“法国蜕皮(French molt)病”[3]。该病1981 年首次报道于美国和加拿大[4-5],随后迅速蔓延到日本、意大利、德国和匈牙利等国家[2,6]。1994 年我国湖北省云梦县和山东省青岛市首次报道发生BFD[7-11]。此后,该病在我国饲养鹦鹉较多的许多省市广泛传播,如湖北、山东、广东等,给我国伴侣鸟贸易和鹦鹉养殖业造成较大经济损失。
APV 属于多瘤病毒科(Polyom aviridae)多瘤病毒属(Polyom avirus),是一种无囊膜的双链DNA 病毒,病毒粒子直径为40~50 mm[4,12],大小约为5 kb[13]。APV 基因组分为早期、晚期基因编码区及非编码区。早期编码区编码1 个大肿瘤抗原(大T 抗原)和1 个小肿瘤抗原(小t 抗原);晚期编码区编码主要结构蛋白VP1 和3 个次要结构蛋白VP2、VP3 和VP4。VP1 蛋白是主要的保护性抗原,而3 个次要蛋白也存在于病毒衣壳中[13-14]。APV 非编码区可能含有复制起始位点和早期及晚期启动增强序列。
目前,该病主要根据临床表现和组织学观察作出初步诊断,而确诊需要进行电镜观察、病毒中和试验或PCR 检测。但现有的检测方法都存在一定的局限性及非特异性。为提高分子生物学方法检测APV 的敏感性和特异性,本研究通过用地高辛(digoxinum,DIG)标记核酸探针,建立了一种快速、灵敏、特异的APV 检测方法,并对鉴定阳性的APV 进行了完整的基因组测序以及遗传特性和进化程度分析。
1 材料与方法
1.1 病毒和病料
APV 参考株:由山东农业大学家禽肿瘤病实验室鉴定、保存。病料:收集山东省某鹦鹉养殖场病死虎皮鹦鹉的肝脏、肾脏、心脏等病料组织,经研磨后采用组织基因组DNA 提取试剂盒(OMEGA),按说明书提取细胞总DNA,-20 ℃保存备用。
1.2 核酸探针制备及灵敏度和特异性检验
利用本实验室保存的APV 参考株VP1基因质粒,根据已经发表的APV 全基因组序列,通过序列比对设计引物(AP-F1:5'-CAGAACATATCGAAATGG-3';AP-R1:5'-TGCAGATATCAAGACTGCC-3'),由上海生物工程有限公司合成探针。按照罗氏公司PCR DIG Probe Synthesis Kit 说明书,合成针对APV 的DIG标记核酸检测探针。将定量好的阳性参考株核酸梯度稀释至103、102、101、100、10-1pg/µL,按照文献[14]方法进行核酸斑点杂交,对制备的探针进行灵敏度检测,同时与普通PCR 进行敏感性比较。
将本实验室分离鉴定和制备保存的禽网状内皮组增生症病毒(REV)cDNA、鸡马立克氏病病毒(MDV)DNA、鸡传染性贫血病毒(CIAV)DNA、禽白血病病毒(ALV)cDNA、鸡呼肠孤病毒(ARV)cDNA、鸡传染性支气管炎病毒(IBV)cDNA、鹦鹉喙羽病病毒(PBDFV)DNA,使用所制备探针,按照文献[14]方法进行核酸斑点杂交,对制备探针进行特异性检测。
1.3 鹦鹉病料APV 核酸检测
参照已发表的APV 全基因组序列[15],通过序列比对,设计合成用于检测APV 的引物(AP-F:5'-GAACATATCGAAATG-3';AP-R:5'-CAGATATCAAGACTG-3')。取2.0 μL 待检病料所提取DNA 模板,加入23.0 μL 的PCR 混合液:2.5 μL 10×PCR buffer,2.5 μL dNTP(2.5 mmol/L),引物AP-F、AP-R(10 μmol/L)各1.0 μL,0.7 μLTaqDNA Polymerase(3.5 U/μL),加双蒸水至23.0 μL。PCR 反应条件为:95 ℃预变性5 min;95 ℃变性30 s,56 ℃退火30 s,72 ℃延伸50 s,共31 个循环;72 ℃延伸10 min。取2.0 μL 上述PCR 产物点样于尼龙膜上,按照文献[15]方法,使用制备探针进行核酸斑点杂交检测。
1.4 阳性样品APV 全基因组克隆测序和序列分析
取斑点杂交检测为阳性的病料组织基因组DNA,参考已发表的APV 全基因组序列[15],设计合成2 对引物(表1),分段扩增2 段相互重叠的片段,用于阳性样品APV 的全基因组克隆测序。按TaKaRa ExTaq酶试剂盒(TaKaRa,Code No.RR001A)使用说明,用这2 对引物分别进行PCR扩增。PCR 扩增产物经1.0%琼脂糖电泳鉴定并将目的条带切下,然后使用E.Z.N.A Gel Extraction Kit(OMEGA,USA)回收纯化DNA;将纯化的DNA 连接到PMD-18T Vector(TaKaRa,Japan)后,转化到大肠杆菌感受态细胞DH5α;挑取单个菌落以Plasmid Mini Kit(OMEGA,USA)提取质粒进行酶切鉴定,阳性克隆送上海生工生物工程有限公司测序。

表1 用于扩增APV 全基因组序列的引物
1.5 序列比对和遗传进化分析
使用DNAstar 软件,将分别测序的两段核苷酸序列进行拼接以获得病毒的全基因组序列,将其与GenBank中已发表的代表性APV参考序列(表2)进行全基因组序列比对,分析分离毒株与不同参考毒株间的核苷酸同源性,并绘制分子遗传进化树。
2 结果与分析
2.1 VP1 基因PCR 扩增和探针标记
以APV-VP1 质粒为模板,用AP-F1 和AP-R1引物扩增VP1基因片段,结果出现与预期相符的731 bp 目的条带。标记的探针从电泳图谱上看,明显滞后于普通dNTP 扩增产物条带(因带有DIG标记的dNTP 分子质量较大,导致电泳速度较低),说明标记成功(图1)。

表2 用于绘制APV 毒株分子遗传进化树的参考株

图1 APV 探针标记结果
2.2 核酸探针灵敏性检测
将APV 阳性对照核酸倍比稀释至103、102、101、100、10-1pg/µL,分别取2 μL 点样在尼龙膜上,使用制备的APV 核酸探针进行斑点杂交。由图2可见,APV 特异性核酸片段为2×103~2×100pg的量时,核酸显色,呈阳性反应;APV 特异性核酸片段为2×10-1pg 的量时及阴性对照,核酸均不显色,呈阴性反应。敏感性统计结果见表3。由图3 可见,当APV 特异性核酸片段在2×105~2×101pg的量时,PCR 电泳结果呈阳性反应;而APV 特异性核酸片段为2 pg 量时及阴性对照核酸,PCR 电泳结果呈阴性反应。结果表明,该探针可检测到2 pg 量的APV 特异性核酸片段,且本检测方法较普通的PCR 检测方法具有更好的敏感性。

图2 APV 核酸探针灵敏性检测结果

表3 APV-VP1 探针的灵敏性检测信号强度统计
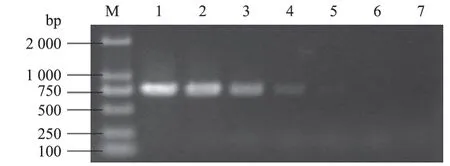
图3 PCR 灵敏性检测结果
2.3 核酸探针特异性检测
将已知背景的REV-DNA、MDV-DNA、CIAV-DNA、ALV-DNA、ARV-DNA、IBV-DNA、PBFDV-DNA 以及APV-VP1 阴性核酸、阳性核酸分别点样在尼龙膜上,使用制备的APV 核酸探针进行斑点杂交。由图4 可见,仅有APV-VP1 阳性核酸显色,呈现阳性反应,而阴性核酸和REV、MDV、CIAV、ALV、ARV、IBV 以及PBFDV 的DNA 均不显色,呈阴性反应,表明合成的核酸探针特异性强,仅识别APV 阳性核酸而不识别其他非APV 核酸成分。

图4 APV 探针的特异性检测结果
2.4 核酸探针病料检测
对收集的8 份病死鹦鹉肝脏、肾脏、心脏等病料组织提取的DNA,用本研究建立的方法进行检测,检出2 份阳性(图5),均来自肝脏样品。

图5 临床病料样品PCR 产物斑点杂交检测结果
2.5 阳性样品APV 全基因组序列测定与进化分析
利用DNAstar 软件,对2 个阳性样品的PCR片段进行了APV 全基因组核苷酸序列测定,并分别命名为APV-SD03 株和APV-SD05 株,全长分别为5 952 bp 和5 956 bp。将序列提交到GenBank,获得的序列号分别是MH712455 和MH712456。
BLAST 和DNAstar 软件分析结果显示,本研究所得到的APV-SD03 株和APV-SD05 株序列与GenBank 中已发表的25 株APV 分离株全基因组序列同源性为99.3%~99.7%。序列比对结果显示,APV-SD03 株共有17 个核苷酸位点的改变,APVSD05 株共有18 个核苷酸位点的改变。统计不同基因组区域碱基变异情况可见,2 株毒株均在大T 抗原区发生了最高的变异率,且APV-SD05 株在T/t抗原区,4 724 位置处有1 个核苷酸替换,并引起了氨基酸V →A 的改变。
对GenBank 收录的25 个APV 全序列以及APV-SD03 株和APV-SD05 株序列,使用MEGA 5.0 软件绘制以全基因组核苷酸序列为基础的APV毒株分子遗传进化树(图6)。在该进化树中,可以将其划分为Ⅰ—Ⅳ家系:家系Ⅰ分离株的地理范围包括美洲、欧洲以及亚洲,其中5 株从鸡分离到,而其他的则从鸟分离到;家系Ⅱ主要为亚洲的日本分离株和中国分离株,都从鸟分离到;家系Ⅲ为单独的大分枝;家系Ⅳ包含了澳洲的新西兰分离株、美国分离株以及2012 年的中国高密分离株,也都从鸟分离到。从进化树推测,分离毒株在物种的特异性上存在一点关系,而在地理分布上没有明显关联。在该研究中,APV-SD03 株和APV-SD05 株位于家系Ⅱ的同一支上,表明两分离株基因组之间在流行病学上相关联;该家系中其他分离株的宿主都是鸟类,且只有FJ385773、APV-SD03 株、APVSD05 株为中国分离株,其他4 株均为日本分离株,可以看出其与日本分离株之间有较近的亲缘关系。2012 年中国高密分离的WF-GM01 株位于家系Ⅳ中,而APV-SD03 株、APV-SD05 株与其基因组序列进行比对后发现有部分氨基酸的突变,从而导致了上述几株毒株不在一个分支上。

图6 APV 毒株分子遗传进化树
3 讨论
APV 是引起鹦鹉类(种、群)发生高死亡率的急性病毒性传染病病原。20 世纪80 年代初,此病首先在美国和加拿大被报道,随后相继在日本、意大利、匈牙利等国家出现。我国首次于1994 年报道该病流行[4-10]。2002—2005 年,有研究[16]证实,我国台湾鹦鹉中存在APV。2008 年后,国内多家宠物鹦鹉养殖场连续暴发由APV 引起的BFD,给我国养鸟业带来重大损失[17]。还有研究[19]显示,不同基因型的APV 在我国共同流行[18]。至今,BFD 仍在我国某些地区散发流行。目前,对于本病的防治尚无较好的控制手段。
目前,APV 核酸检测主要基于建立的PCR 法。该方法虽灵敏度高,但有时会出现非特异性反应,一般需要将PCR 产物回收后连接到相应载体上,经过测序才可作出最终判断,这就大大增加了检测的时间和成本。因此,亟需建立一种更加快速、准确、易操作的检测方法。DIG 标记DNA 探针杂交检测法具有特异性强、灵敏性高、操作方便、检测时间短、性质稳定等优点,是目前常用的分子生物学诊断技术。本研究建立了一种针对APV 的特异性核酸探针斑点杂交检测方法,并通过相应试验验证了其可以检测到2 pg 量的APV 核酸,且只对APV 核酸呈现阳性反应,而对REV、MDV、CIAV、ALV、ARV、IBV 以及PBFDV 的DNA 均呈现阴性反应,证明该APV 的核酸探针斑点杂交检测方法具有较高的灵敏度和较强的特异性。
本研究利用制备探针对发病鹦鹉病料进行APV 检测,共检出2 份阳性样品,并对其进行了全基因组序列测定及遗传进化分析,结果发现2 株毒株与国内外已发表的25 株APV 全基因组核苷酸同源性高度一致,为99.3%~99.7%。该序列分析结果也与之前Rott 等[20]、Phalen 等[21]和Johne等[22]研究报道的结果一致。APV 序列分子结构方面,通过序列比对,分析其碱基变异情况,可知T抗原变异率最高,推测这可能与APV 的宿主范围广泛有关。APV 全基因组序列绘制的遗传进化树显示,27 株APV 共划分为Ⅰ—Ⅳ家系。本研究的APV-SD03 株和APV-SD05 株都位于Ⅱ家系的同一分枝上,表明两分离株在流行病学上相关联。从整体进化树可推测,分离毒株在种属特异性上存在一点关系,而在地理位置间没有显著关系。
我国是一个候鸟迁移和家禽养殖大国,因此有效控制鸟类的APV 感染非常重要。本研究建立了一种灵敏度更高、特异性更强的PCR 产物斑点杂交检测方法,并对鉴定阳性的毒株进行全基因组序列测定及遗传进化分析,从而加深了对APV 进化途径的了解,对于预防和控制APV 感染具有重要意义。
猜你喜欢
青少年科技博览(中学版)(2022年10期)2023-01-07
作文周刊·小学二年级版(2022年20期)2022-05-05
科学大观园(2022年2期)2022-01-23
意林·全彩Color(2019年6期)2019-07-24
小学生作文(中高年级适用)(2017年4期)2017-07-10
现代检验医学杂志(2016年3期)2016-11-15
动物医学进展(2015年10期)2015-12-07
物理实验(2015年9期)2015-02-28
特产研究(2014年4期)2014-04-10
郑州大学学报(理学版)(2014年3期)2014-03-01
