
药物-药物共晶的研究进展
2021-03-06 02:58:02孙晶晶贾丽娜林波王艳龚俊波
化工学报 2021年2期
孙晶晶,贾丽娜,林波,王艳,龚俊波
(1 天津大学化工学院,化学工程联合国家重点实验室,天津300072; 2 国家工业结晶工程技术研究中心,天津300072)
引 言
传染病、艾滋病、癌症、糖尿病和心血管疾病等复杂疾病已经成为制约人类平均寿命提高的重要因素,由于单一疗法(即针对特定受体)在许多复杂疾病的治疗中效果不显著,联合用药已成为一种被普遍认可的药物开发策略,一般通过复方制剂或同时服用多种的方式实现联合用药。复方制剂指两个或者两个以上的药物活性分子(API)结合在单剂量中,不仅具有比单独给药更有效的治疗效果,还具有减少处方数量和管理费用、增加患者的依从性等优势[1],近几年来备受关注。然而,根据最近临床中使用的复方制剂,由于配方阶段存在的稳定性差、母体原料药之间溶解度差异大和不相容等问题,优势被大大减弱。因此,有必要开发新技术和新方法以促进联合用药的发展[2]。药物-药物共晶作为联合用药的一种新方法,不仅不会影响药物本身的活性,而且可以显著改善药物的物理化学性质,有望克服传统联合用药中存在的问题[3]。
在美国食品和药物管理局(FDA)引入关于共晶的指南,欧洲药物管理局(EMA)发表关于共晶使用的文件后,共晶被赋予“新的活性物质”(NAS)的地位[4],而药物-药物共晶作为共晶中的一种,符合专利的新颖性、创造性和实用性要求。但目前关于药物-药物共晶的研究大多数停留在共晶的筛选、表征及部分构效关系研究方面,临床应用较少。药物-药物共晶比复合药物和普通类型的共晶具有更高的不可预测性,这将大大限制药物-药物共晶的工业化和在临床中的使用。
本文将以近些年药物-药物共晶的案例为基础,介绍目前药物-药物共晶的形成、溶解和代谢机理,以及如何进行药物-药物共晶的设计和预测,为药物-药物共晶的进一步研究提供指导。
1 药物-药物共晶的概念及优势
药物共晶是指在同一晶格中活性药物分子(API)和共晶形成物(CCF)按照固定的化学计量比通过非共价键和非离子键的相互作用构成的共晶[5]。药物-药物共晶是药物共晶的一个子集,其中共晶形成物也是一个具有独立药物活性的生物活性分子。目前,药物-药物共晶被视为固定剂量的组合产品,是通过非共价相互作用而结合在一起的超分子复合物。由于共晶不涉及原料药化学结构的改变,与新的化学药物相比,其开发周期大大缩短,为改善新药开发费时费力的现状提供了一个新的思路[6]。药物-药物共晶作为联合用药的新药制剂的开发方法,将为同时治疗靶向性疾病或多种疾病开辟一条新的途径。
大多数药物分子在药代动力学和物理化学性质(溶解度、溶解速率、引湿性、膜渗透性、化学稳定性等)方面存在不足,这些性质在很大程度上会影响药物活性成分在临床上的治疗效果。药物-药物共晶为改善药物的物理化学性质提供了一种新的思路,将两种物理化学性质互补的药物制备成共晶,不仅可以显著地改善两种药物物理化学性质的不足,而且实现了联合用药的理念,具有更好的疗效。例如,二甲双胍和格列齐特都是有效的口服降血糖药,并且这两种药物在控制血糖和血脂指数方面具有协同作用,一般联合使用[7]。但二甲双胍原料药具有很强的引湿性,在生产过程中必须特殊处理和使用封闭包装系统,这会极大地增加药物成本。根据生物药剂学分类系统,格列齐特是一种BSC Ⅱ类药物,溶解度差,很大程度上限制了其生物利用度。Putra等[6]制备了二甲双胍和格列齐特的药物-药物共晶,该晶体存在一个亲水性的通道结构,高引湿性的二甲双胍被低溶解度的格列齐特包裹在里面,如图1所示。与格列齐特原料药相比,药物-药物共晶具有更好的溶解性和溶解速率,二甲双胍的高引湿性问题也通过共晶的形成得到显著改善。

图1 二甲双胍−格列齐特多组分晶体结构示意图[6]Fig.1 Schematic diagram of the multi−component crystal structure of metformin−gliclazide[6]
药物稳定性对药物的有效性和安全性会产生重要影响,API 转化为非活性代谢物会降低其治疗效果,甚至产生毒性。例如维生素D2和维生素D3(VD2和VD3)广泛应用于食品、药品等行业,目前大部分生产都在中国完成,全球市场每年消耗近100 t[8]。但由于维生素D 容易发生降解,故在储存和运输过程中可能会失去活性,不明的降解杂质会对人体健康造成不可预测的风险。Mei 等[2]制备了VD2和VD3的两种药物-药物共晶的多晶型(A 和B),结构如图2所示,但两种多晶型化合物的理化稳定性存在显著差异,与单独的VD2和VD3相比,A 型具有优越的物理化学性质优势,稳定性测试结果如图3所示。这项工作表明,药物-药物共晶有利于开发更稳定的维生素产品,但药物-药物共晶的多晶型现象也是影响药物稳定性和疗效的重要因素。

图2 VD2和VD3共晶的设计与合成[2]Fig.2 Design and synthesis of co−crystals of VD2and VD3[2]

图3 不同固态形式的维生素D3在储存期间测定值变化[2]Fig.3 Changes in vitamin D3 assay values during storage[2]
多晶型是指同一化合物在固态中存在一种以上的晶体结构。尽管单药物多晶型广泛存在,例如巴比妥、岩藻糖、卡马西平、乙胺嘧啶、咖啡因等,但药物−药物共晶中的多态性还是相对较少[9]。药物−药物共晶多晶型是指两个API 之间形成的2 种或2种以上具有不同晶体结构的共晶[10]。与单组分药物的多晶型分类相类似,根据API 分子在晶格中药物分子之间的连接方式、空间排列方式、分子构象等方面的不同,药物−药物共晶多晶型也可分为合成子多晶型(synthon polymorph)、堆积多晶型(packing polymorph)、构象多晶型(conformational polymorph)以及互变异构多晶型(tautomeric polymorph)等[10]。多晶型会影响药物的性质,如溶解度、稳定性、生物利用度等,故多晶型的重要性及其在药物开发中的普遍性是公认的。Mei 等[11]报道了三种1∶1(摩尔比)的氨苯砜−黄酮共晶的多晶型(A,B 和C),在pH 为6.8 的缓冲介质中,A 型比C 型的固有溶解速率高出约8 倍。但目前报道的药物−药物共晶的多晶型往往是在制备共晶的过程中意外被发现的,没有形成一个科学的筛选系统。聚合物诱导的异核作用提供了一种从单一溶剂中获得多种成核条件的方法,因此非常适合开发共晶多晶型[12]。当通过形成共晶的方式来调整具有理想性质的原料药时,必须考虑形成共晶多晶型的可能性。与单组分晶体类似,固体形态的筛选和药物的系统表征和评价是药物−药物共晶发展的重要步骤。
药物−药物共晶不仅能够改善原料药的物理化学性质,还可以发展联合疗法,预防多药耐药性,协同增加药物的作用,减少副作用等[13−14]。原料药拉米夫定和齐多夫定均是有效的治疗艾滋病毒的药物,拉米夫定可以抑制两种类型(Ⅰ型和Ⅱ型)的艾滋病毒逆转录酶[15],而齐多夫定通过阻止艾滋病毒的复制,进而延缓疾病的发展,在临床治疗中联合使用这两种药物具有更好的治疗效果[16]。Desiraju等[17]报道了两种抗艾滋病药物拉米夫定和齐多夫定的药物−药物共晶,晶体结构如图4(a)所示,由于该药物−药物共晶同时具有这两种药物的药理作用,可能成为治疗艾滋病更有效的药物。维甲酸(DIM)是一种广泛用于预防晕车的药物,市售DIM 被称为多组分晶体,由苯海拉明和8−氯茶碱两种药物组成,二聚体结构如图4(b)所示。苯海拉明是H1组胺受体的拮抗剂,但有嗜睡的副作用。8−氯茶碱是一种兴奋剂,可以克服苯海拉明的副作用[18]。磺酰脲类药物通过刺激胰腺内胰腺细胞的胰岛素分泌和降低肝脏清除率发挥作用,而二甲双胍可提高胰岛素敏感性,从而降低非胰岛素依赖型糖尿病(NIDDM)中普遍存在的胰岛素抵抗。二甲双胍控制血糖的效果与磺脲类药物相似,由于它们的作用方式不同,在临床治疗中通常作为药物组合使用[19−20]。这两种药物的联合治疗不仅可以改善血糖控制,而且在某些情况下也会使总体药物剂量和不良反应最小化[7]。Gong 等[21]报道了含甲苯磺丁脲和二甲双胍的药物−药物多组分晶体,晶体结构如图4(c)所示,与二甲双胍相比,其吸湿性降低,加速稳定性增强;与甲苯磺丁脲相比,提高了溶解度和溶出度。药物−药物多组分晶体作为联合用药的新方法,有望发挥联合用药的优势,成为治疗Ⅱ型糖尿病的有潜力的替代品。
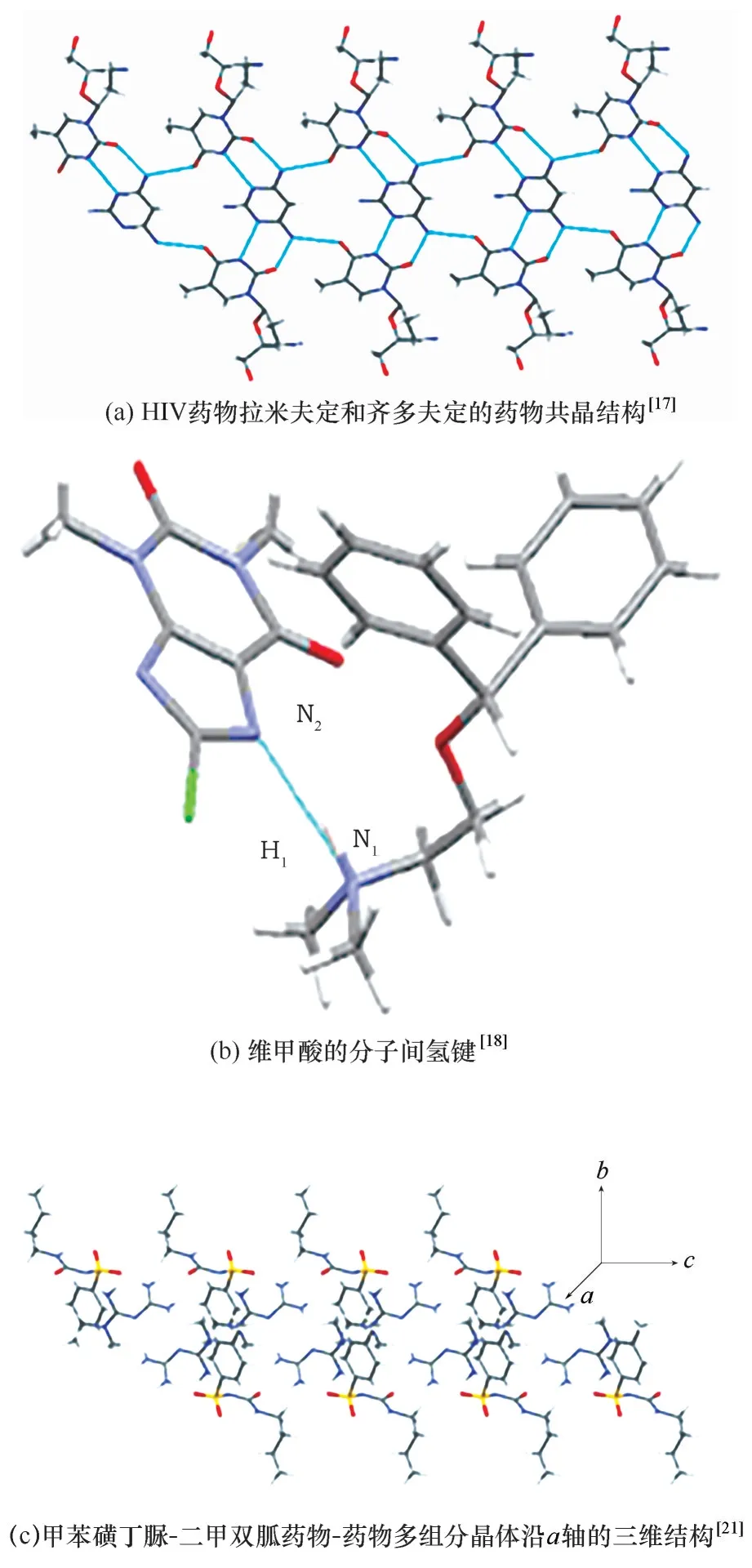
图4 药物−药物共晶的晶体结构[17−18,21]Fig.4 The crystal structure of the drug−drug co−crystals[17−18,21]
基于晶体工程的药物−药物共晶使联合用药得到了发展,药物−药物共晶不仅具有药物共晶中改善药物物理化学性质的优势,而且体现了联合用药的理念,提高了患者的依从性,在多种疾病治疗过程中减少了药物负荷,提高了成本效益。此外,药物−药物共晶还可延长药物的专利保护期限,故受到制药企业的广泛关注,相关专利申请量也在快速增加。开发药物共晶的策略,尤其当所有组分都是原料药时,药物−药物共晶显示出比其单独作用之和更好的效果,具有广阔的市场前景。
2 药物−药物共晶的形成、溶解和代谢机理
2.1 药物-药物共晶的形成机理
药物−药物共晶的制备方法可分为固体法和溶液法。固体法主要是指使用很少溶剂或不使用溶剂的方法,如研磨法、热熔挤出等。固体法的共晶形成机理主要是从反应物相、中间相和产物相之间的宏观转变来考虑的。固态方法制备共晶的机理尚不明确,主要分为三种机制:分子扩散[22]、共熔体的形成[23]和无定形重结晶[24],这三种不同机制的中间相分别是气体、液体和非晶态的固体,其共同点是中间相的反应物分子相对于起始晶型具有更高的迁移率和更高的能量[25]。溶液法主要是指涉及大量溶剂的制备方法,如冷却结晶、蒸发结晶等,需要分离操作将结晶产物从母液中分离出来。在溶液中关于共晶形成机理的研究主要停留在热力学以及宏观形成机理的探索,如相图、溶度积等,在分子层面关于共晶的形成机理研究较少,研究分子层面共晶的成核以及晶体的生长调控等将有助于更好地理解溶液中共晶的形成机理,为共晶的设计和开发提供更多的理论基础。
若一种或者两种反应物在固态时有明显高的蒸气压,这两个固体反应物之间很可能通过分子扩散的方式形成共晶,新的可用来反应的表面的形成有利于增强反应物晶体之间的分子扩散[22],机理如图5(a)所示。通过分子扩散形成共晶的最大缺点是共晶的形成受到反应物晶体上可用来形成共晶的表面的面积限制。例如当反应物的晶体结构中有广泛的氢键网络,这种氢键网络将会阻止分子的表面扩散。在分子扩散介导的共晶形成体系中,可通过暴露新的反应物表面和加强两个固体间的混合来提高反应速率[22]。机械化学研磨在这类系统中的作用主要是通过去除产物和产生缺陷等方式来增加新的可以用来反应的表面,增强混合反应物的表面扩散。
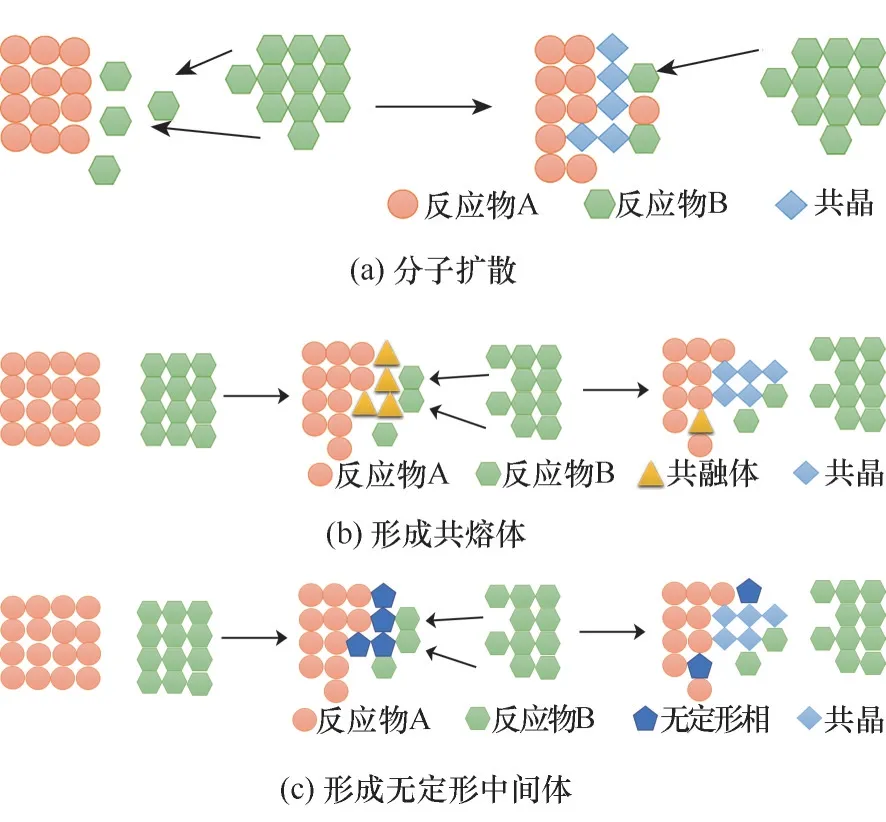
图5 共晶形成机理示意图Fig.5 Schematic of cocrystal formation mechanism
在共熔体液相介导的研磨法制备共晶的过程中,中间液相促进了固体共晶产物的形成,随后成为共晶产物的一部分,并随着反应接近完成而逐渐消失,机理如图5(b)所示。Davey 等[23]在二苯胺和二苯甲酮共晶的形成过程中研究了亚稳的共熔体液相的形成,通过对二苯胺和二苯甲酮两个宏观单晶之间界面的微观观察,界面开始熔化直到大部分物质在室温下转化为液体,随后共熔体成核导致整个熔体凝固形成固态共晶。在上述过程中,研磨过程起了很大的作用,研磨使新反应物表面暴露,通过诱导共熔体液相形成共晶核,促进共晶的形成[26]。
若两个固体不易挥发且分子间通过强相互作用(如氢键)连接,共晶的形成机理最有可能通过非晶态中间体进行,机理如图5(c)所示。由于大量的药物化合物不易挥发且存在可以形成氢键的官能团,通过干磨法制备的药物−药物共晶大多是以非晶态中间体为介导形成共晶的。例如,Rodríguez−Hornedo等[27]以卡马西平和糖精为模型物,通过干法研磨发现,在制药过程中产生的非晶相导致共晶的产生。在此基础上,他们针对药物−药物共晶的形成机理进行了深入的探索,在略低于混合物的预期玻璃化转变温度的条件下进行研磨,将产生一个非晶态相,在室温下缓慢地进行共结晶将形成共晶。然而在室温下研磨两种反应物会导致部分非晶态和共晶的形成,通过机械化学搅拌产生具有高能量和高分子流动性的非晶态中间体,之后非晶态转变为共晶。非晶相的转化在低温下受到抑制,并且在接近非晶相玻璃化转变温度下得到促进,该温度可由单个共晶成分的玻璃化转变温度近似得到。
以非晶态为中间体的共晶的形成取决于非晶态中间体的玻璃化转变温度,使用增塑剂(如水),可以在较低温度下促进共结晶,提高共晶的形成概率。除了作为增塑剂外,水还可以在潮解条件下促进固体混合物生成共晶,在这种情况下,通过三个步骤形成共晶,包括吸湿、反应物溶解和共晶的成核和生长[28]。结晶水对固态研磨反应过程的影响是不同的,水合物的稳定性越低,越有利于共晶的形成[29]。
液体辅助研磨的作用机理尚不清楚,目前有两种观点,一些研究认为少量的液体可以作为反应的润滑剂[3],另一些研究认为液体提供了一种增强分子扩散的介质[30]。与溶液结晶相比,液体辅助研磨实验中涉及的液体量通常很少,这表明单个组分的溶解度对于通过研磨能否形成共晶不是决定性因素,故液体辅助研磨可以有效避免共晶各组分间溶解度差异的问题,提高共晶的形成概率,提供难以从溶液中获得的共晶产品[31]。
目前已用溶液法制备了大量的共晶,但对于其机理的报道很少。近几年,研究人员对溶液法中共晶的形成机理进行了初步的探索。Hao 等[32]以间甲酚和尿素为模型物揭示了溶液中分子识别和自组装的机理,在溶液中通过降低温度得到间甲酚和尿素共晶,形成过程分为三个部分,首先形成间甲酚和尿素的二聚体,其次是共晶的成核,最后是共晶的生长。但对于其基于溶液法提出的上述机理的普适性,有待进一步考察。在此基础上,Sun 等[33]对溶液中单组分结晶和共结晶的竞争机制进行了深入的研究,在溶剂缓慢蒸发条件下,溶剂中的分子可能以两种方式形成晶体,一种方式是相同的分子结合形成单独的沉淀相,另一种方式是不同的分子结合形成共晶沉淀相。在溶剂缓慢蒸发的条件下,若共晶在溶剂体系中溶解度最小,则共晶最先达到过饱和状态进而优先析出。在相反的情况下,则单组分晶体首先析出。在不同的条件下,通过调节共晶和单组分晶体的析出顺序,有利于制备得到药物−药物共晶产品。共晶形成机理的研究已经拉开序幕,但更完善的共晶形成机理还需要研究人员长时间的共同努力。
2.2 药物-药物共晶的溶解机理
共晶的溶解度取决于两个重要参数,即晶格中分子间相互作用的强度和共晶组分的溶剂化。Maheshwari 等[34]报道了共晶体在溶解介质中的增溶,包括两个主要步骤:(1)溶质分子从共晶晶格中的释放;(2)释放分子的溶剂化。因此,为了增加溶解度,可以降低晶格能或增加溶剂亲和力。共晶对这两种因素都有不同程度的影响。在溶剂化阻力小时,或当溶剂−溶质相互作用与溶剂−溶质相互作用匹配时,如在理想溶液中,晶格能的大小将决定溶质在溶液中的溶解度。另一方面,由于药物的疏水性,溶剂−溶质相互作用(疏水性)所施加的限制使观察到的溶解度低于晶格能所确定的溶解度,这时溶剂化作用将对共晶的水溶性起决定性作用,大多数疏水性药物的共晶都表现出这种特性[35]。
“弹跳−伞降”现象对改善API 的溶解性和溶解速率起着关键作用。Babu 等[36]指出,药物共晶的溶解可以用“弹跳−伞降模型”[37]来解释,药物−药物共晶作为一种特殊的药物共晶,其溶解机理也符合“弹跳−伞降模型”,如图6 所示。药物−药物共晶中不同的API 之间的氢键在生物介质中于短时间内(几分钟到一个小时)被破坏,从而不同的API 被分开[38]。随着共晶的解离,药物−药物共晶中水溶性更高的API从晶格中扩散到生物介质中,此时,共晶中水溶性较低的API也随之脱离晶格在生物介质中变得过饱和,并形成松散聚集的簇。这种纳米分子团簇类似于非晶态药物相,即具有短程有序性,但缺乏长程周期性,这种通过共晶解离生成的非晶态药物形式可以在药物溶解度上表现出与分散在聚合物基质中的非晶态药物相同的尖峰,因此,“弹簧”效应是通过将共晶解离成无定形的药物形式来实现的。这些无定形簇转变为稳定的结晶相或晶体生长将是一个缓慢的过程,非晶态到晶态的转变将被与药物一起存在于胃中的添加剂、聚合物、赋形剂、增溶剂等所抑制。高能非晶富相溶液将转变为药物的亚稳态晶型(具有更高的溶解度),然后根据Ostwald 的阶段定律最终转变为稳定的热力学晶型[36,39]。Ostwald 的阶段定律扩大了亚稳态宽度,故使“降落伞”效应时间延长,维持峰值溶解度或“降落伞”效应的时间足够长(120~300 min)可提供高剂量溶解度[40]。药物−药物共晶的“弹跳−伞降模型”可以有效提高难溶性药物的溶解度、溶出速率和生物利用度等。Shi 等[41]制备了黄芩苷(BE)与烟酰胺(NCT)共晶,共晶的体内溶出曲线表现出“弹跳−伞降”现象,如图7所示,显著提高了BE 的溶解性和生物利用度。与BE 相比,大鼠体内的共晶的峰值血药浓度(Cmax)高2.49 倍,曲线下面积(AUC)高2.80 倍。这种口服生物利用度的显著提高甚至超过了BE 纳米晶。与制备复杂耗时的纳米晶相比,共晶可能为难溶性药物的性能改善提供一种有效的方法。
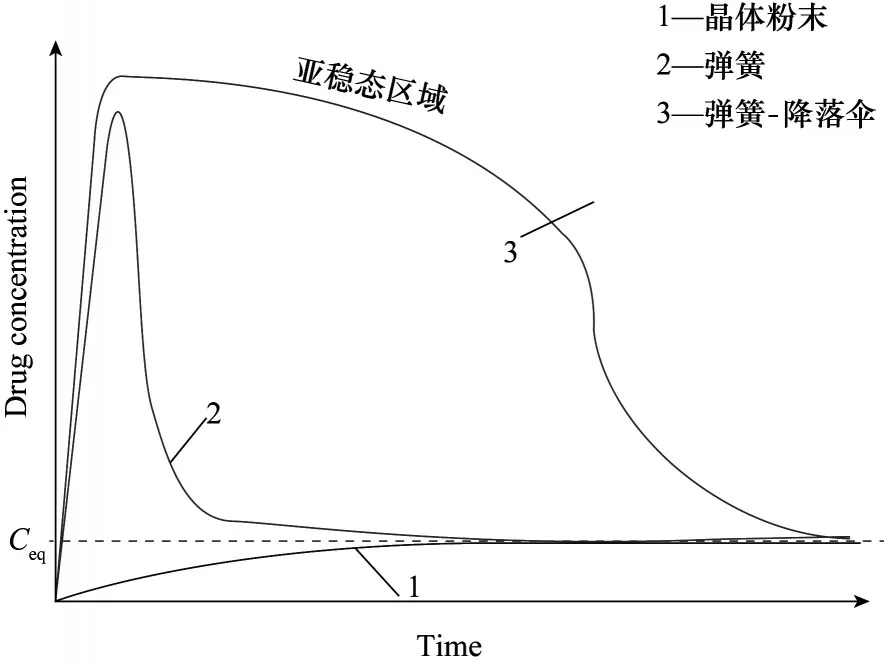
图6 “弹跳−伞降模型”示意图[36]Fig.6 Schematic diagram of“spring and parachute model”[36]
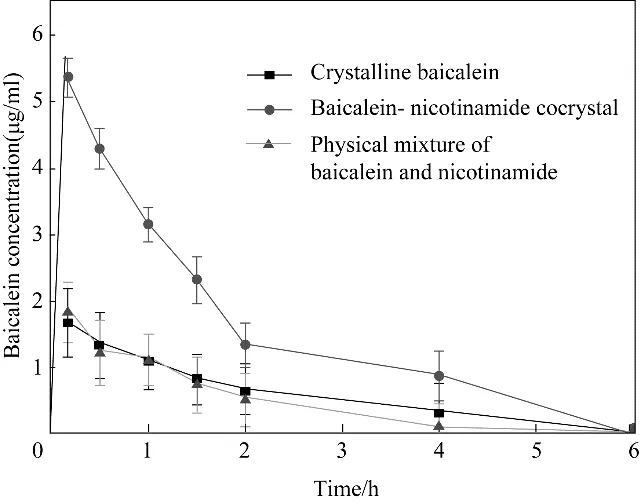
图7 大鼠经口灌胃给药后,结晶态BE、BE和NCT的物理混合物[1∶1(mol)]和BE−NCT共晶体的血浆BE浓度−时间曲线[数据表示为平均值±SD(n=6)][41]Fig.7 Plasma BE concentration−time curves of the crystalline BE,the PM of BE and NCT[1∶1(mol)],and the BE−NCT cocrystal after oral gavage administration to the rats[Data are expressed as means±SD(n=6)][41]
共晶在解离时的另一种途径是释放出亚稳态纳米晶形式的药物,而不是无定形团簇。无论哪种方式,高熵粒子和较大的接触面都会导致晶体更快的溶解[36]。如果无定形状态直接转变为稳定的晶体形式,而不经历亚稳多晶型的中间体状态,药物只会表现出弹簧效应,如图8 所示。例如开发中的非离子化原料药的戊二酸共晶的溶解过程[42],共晶达到表观溶解度峰值(仅弹簧效应),但随后由于直接转化为稳定的药物晶型而浓度下降(无降落伞相)。然而卡马西平−糖精共晶在溶解过程中的浓度分布与药物卡马西平相似,并没有表现出弹簧效应[43]。
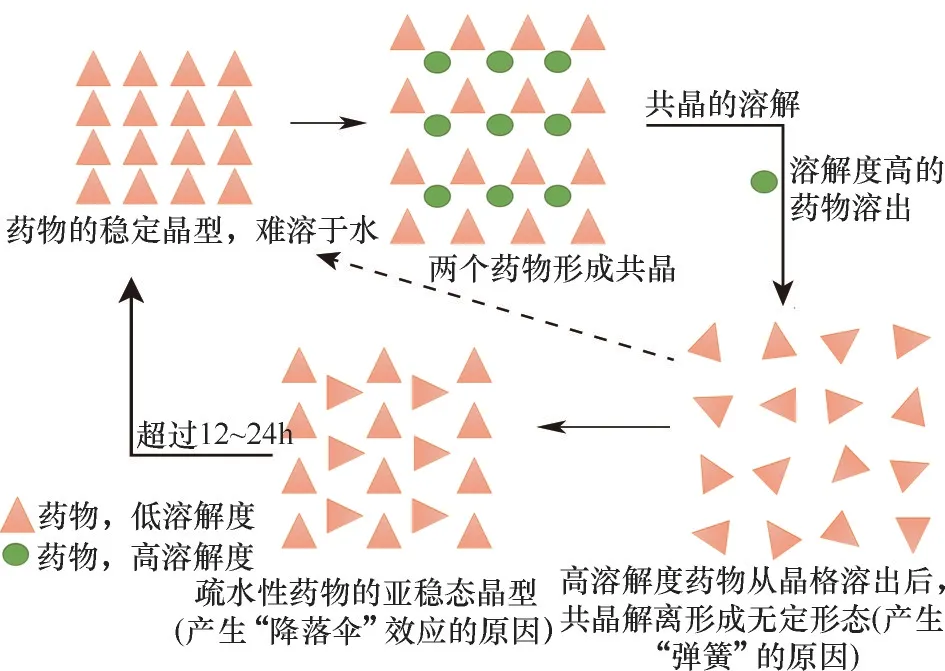
图8 药物−药物共晶的溶出机理[36]Fig.8 Drug−drug co−crystal dissolution mechanism[36]
共晶通过高的瞬时过饱和度来提高药物的溶出度,故其溶出过程必须满足两个条件:药物亚稳态形式的产生和维持足够长时间的高的药物浓度[44]。只有在能够阻止难溶性药物活性组分的沉淀时,高溶性共晶才有较高的溶出率和较好的生物利用度。“弹簧和降落伞”现象对改善API 的溶解性和溶解速率起着关键作用,过饱和给药系统可以为制药行业不断遇到的难溶性药物提供解决方案。
2.3 药物-药物共晶的代谢机理
目前,药物−药物共晶在代谢机理方面的研究较少,但对代谢机理的深入研究是药物−药物共晶从实验室阶段进入临床治疗的重要条件。心力衰竭(HF)的主要药物策略是从阻断肾素−血管紧张素−醛固酮(RAAS)和抑制脑啡肽酶两方面考虑。两种治疗策略均可降低发病率和死亡率,但也带来相当大的不利影响[45]。新一代的HF 药物商品名Entresto[46](诺华公司,研究名称为LCZ696),是包含沙库比曲、缬沙坦二钠和水分子的药物离子共晶体。Entresto 是一种新的血管紧张素受体−脑啡肽酶抑制剂(ARNI),代表了一类新型的治疗心衰的药物,其多模式作用包括抑制脑啡肽酶和阻断血管紧张素Ⅱ1型(AT1)受体。沙库比曲是一种脑啡肽酶抑制剂,而缬沙坦是一种血管紧张素受体阻滞剂。血管紧张素Ⅱ是引起血管收缩的激素,能导致高血压和心脏劳损,脑啡肽酶抑制剂有降低血压的作用。沙库比曲与缬沙坦的组合通过LBQ657(前药缬沙坦的活性代谢产物)抑制脑啡肽酶和选择性阻断AT1受体来抑制血管紧张素Ⅱ的作用。这两种药物的组合不但能够抑制过度活跃的肾素−血管紧张素−醛固酮系统(RAAS)的有害作用,而且可以增强保护性神经激素系统(利尿钠肽系统)的功能[47]。
3 药物−药物共晶的设计及预测
3.1 药物-药物共晶的设计
开发药物−药物共晶需要选择合适的药物组合,综合考虑多个因素,包括治疗效果、溶解度差异、药物−药物相互作用以及两个药物分子或多个药物分子之间形成共晶的概率。对于形成药物−药物共晶的药物组合的筛选,一般分为两步,首先在药学上是可接受的条件下,判断所选药物组合在药理作用上是否能够产生协同作用,其次研究药物组合是否能够形成共晶。
药物组合筛选的第一阶段一般可以从两个角度出发:(1)选择联合治疗同一类病的药物。例如替米沙坦和阿替洛尔均是治疗高血压的一线药物,在临床治疗中可联合使用,可以显著降低单药难以治疗的高血压。Chadha等[48]通过机械研磨的方法制备了替米沙坦−阿替洛尔共晶,该共晶与物理混合物相比,具有更好的降血压活性。(2)选择添加一个特殊用途的药物,如降低副作用或者预防相关疾病的并发症等。例如奥氮平是最广泛使用的抗精神病药物之一,但它存在体重增加、阳痿、Ⅱ型糖尿病和血糖水平升高等多个代谢副作用,为避免奥氮平的代谢副作用,Thakuria 等[49]选择那格列奈和奥氮平的药物组合制备共晶,那格列奈是Ⅱ型糖尿病的治疗药物,与奥氮平制备的药物−药物共晶有望显著减少奥氮平的副作用。基于上述方法选择制备药物−药物共晶的药物组合还应确保选择的组合不影响每种药物的稳定性。
制备药物−药物共晶的药物组合除了要考虑上述的药理学的因素,还应考虑所选的药物组合之间能否形成共晶。目前,还没有可以准确预测药物−药物共晶形成的方法,但关于药物共晶的预测已有许多报道,而且能成功地应用于药物−药物共晶形成的预测[50]。由于共晶的形成受到多个重要参数的影响,因此选择合适的共晶形成物对于成功地制备出共晶至关重要(在药物−药物共晶中,其中一种原料药充当共晶形成物)。关于共晶形成物的筛选可以通过实验或理论预测进行,理论预测的方法可作为快速筛选共晶形成物的工具,但由于准确度较低,仅可用来初步评估适用于共结晶过程的共晶形成物。

图9 代表性的超分子合成子[51]Fig.9 Representative supramolecular synthons[51]
关于共晶形成物的选择,在理论预测方面通常是基于合成子的方法,该方法通过特定的分子片段来建立“超分子合成子”。超分子合成子是通过互补官能团之间的非共价键所构成的,常见的超分子合成子如图9[51]所示。这个概念表明需要详细了解给定API 中存在的官能团的超分子化学,这是设计共晶的第一步。之后通常会对剑桥结构数据库进行一次调查,以识别一系列可以与给定API 中的官能团形成常见的(即稳定的)超分子合成子的相关结构。此后,选择满足共结晶体系中的官能团互补规则的潜在的共晶形成物[52]。一般来说,可以利用一系列常见官能团中的超分子合成子的层次结构。研究结果显示[53−56],某些官能团,如羧酸、酰胺和醇羟基特别容易形成超分子杂合子。合成子的方法表明,药物和共晶形成物上的特定官能团将在共晶的形成中发挥重要作用。但合成子的方法也存在一些缺点。由于超分子本身可能不能堆积成有序的晶体结构,因此它不是定量的。另外,该方法没有考虑诸如API或共晶形成物内存在的不同官能团之间的竞争或供体和受体周围的空间密度之类的因素[57]。
3.2 药物-药物共晶的预测
设计活性药物成分(API)的多种固体形式(如多晶型、盐、共晶、水合物和溶剂化物)常用于控制和调节API的理化和药代动力学性质,如稳定性、可压缩性、过滤性、溶出度、生物利用度、压片等。成盐是最常见和最有效的方法,但只能用于提高可电离药物的溶解度和稳定性[58],共晶为缺乏可电离官能团的原料药改善其物理化学性质提供了另一条思路。根据FDA《药物共晶监管分类指南(2018)》[5],共晶是指在同一晶格中包含两种或以上不同分子按照固定的化学计量比通过非离子键和非共价键结合形成的晶体材料,而盐则是由金属或类金属基团取代部分或全部酸性氢而形成的离子或电价键的结晶化合物。从监管的角度来看,共晶不需要注册新的活性物质或化学实体,而在药物盐的情况下需要注册新的活性物质或化学实体。一般根据质子的转移程度来区分盐和共晶,在实验中常通过单晶X−射线衍射、固态核磁共振、X 射线光电子能谱、固体红外和拉曼图谱等方法进行判断[59],在理论预测方面,ΔpKa规则可用于对多组分晶体的初步判断[5]。一般而言,如果API 及其共晶形成物的ΔpKa(pKa(碱的共轭酸)−pKa(酸))>1,则质子会大量转移,从而导致电离并可能形成盐。另一方面,如果API 及其共晶形成物的ΔpKa(pKa(碱的共轭酸)−pKa(酸))<1,则质子转移将不充分。如果满足此条件,则应将API 和共晶形成物构成的化学物质归类为共晶。5−氟胞嘧啶和5−氟尿嘧啶之间的ΔpKa=−3.0,Ellena 等[60]利用ΔpKa规则成功制备出包含5−氟胞嘧啶和5−氟尿嘧啶的药物−药物共晶。但利用ΔpKa规则预测共晶的形成存在许多问题,该标准并不总是适用,因此需要与其他的预测手段相结合进行共晶预测。
迄今为止,文献报道的共晶形成物的筛选计算方法大多是基于热力学的方法,一些参数经常被用于计算预测共晶的形成或共晶形成物的筛选,其中包括相互作用能的计算[61−62]、静电势[63]、API 和共晶形成物之间的分子互补性[64−65]、API和共晶形成物的晶体能量分布[66]和氢键倾向[67]等。Price 等[68]通过晶格能计算筛选可以形成热力学稳定的共晶的共晶形成物,如果共晶的晶格能低于单个组分的晶格能之和,则共晶相被称为热力学稳定相。共晶只有在热力学上比共晶组分更稳定,才有望形成共晶。Velaga 等[69]根据Hansen 溶解度参数预测,指出药物与共晶形成物的混溶性可以指导共晶形成并进行共晶形成物的筛选。结果表明,药物和共晶形成物的可混溶性是能够形成共晶的必要条件。因此,使用溶解度参数预测共晶组分的可混溶性可对潜在的共晶形成物进行初步的筛选。McCabe 等[62]通过比较共晶的相互作用位配对能与两个纯组分的相应能量(ΔE)提供了一种评估共晶体形成概率的方法,在大多数情况下,计算发现实验观察到的命中率具有较大的ΔE 值,与基于COSMO 的虚拟共晶筛选方法进行比较,发现这两种方法之间差别不大。Fabian[67]通过对CSD 中已知共晶的统计分析,确定了影响共晶形成的分子性质。形成共晶的分子的形状和极性趋于相似,而在氢键供体和受体的数值“不平衡”方面,没有体现出互补性,这种现象为合理设计共晶提供有用的定性指导和半定量预测模型的出现提供了基础。但关于共晶形成预测的理论方法目前仅停留在单因素预测,预测的准确性还有待提高。
热分析可以很容易地检测到多组分反应物分子(API 和共晶形成物)形成的新相(如共晶),可用于共晶形成物的筛选[70]。最近,Wesolowski等[71]使用差示扫描量热分析(DSC)作为快速筛选工具,识别出15 种不同的苯二氮卓共晶(化学计量比为1∶1)。DSC 可作为一种工具,通过差示扫描量热结果和熔融相图识别能够与API 分子形成共晶的共晶形成物[72−73]。以特定化学计量比获得的原料药和共晶形成物的二元混合物的DSC 数据有助于确定固体形式的性质,当DSC 显示出熔点低于或介于或大于单个组分熔点的单一吸热时,二元系统很可能形成共晶体,与X 射线粉末衍射(XRD)分析相结合可以证实新共晶相的形成[74−75]。此外,如果DSC 热分析图显示连续的多个峰,则可能表明共晶的形成。当存在两个连续的峰值时,第一个熔化峰对应于低共熔混合物的熔点,随后是共晶的熔化(用第二个吸热曲线表示)。两个或多个峰也可能是共晶的多态性转化的信号。如果在DSC 中存在三个连续的吸热,第一个表示低共熔混合物的熔化,第二个表示多余成分(药物或共晶形成物)的熔化,第三个是吸热下共晶的最终熔化[70,72]。
相图可用来识别不同的固相,这些固相可以在任何药物和共晶形成物之间形成。相图可分为二元相图(API−共晶形成物)或三元相图(API−共晶形成物−溶剂)。包含不同化学计量的药物和共晶形成物的混合物的DSC 可用于构建任何药物和共晶形成物的二元相图。这些二元相图可以用来检测给定系统的形成共晶的区域。通常使用从热分析方法(例如DSC 分析)获得的数据点来构建二元相图。并将DSC 热分析图中第一个吸热的起始温度选择为固相线。选择第二个吸热峰温度作为液相线来构建相图。API 和共晶形成物的熔融行为(全融/非全融)决定了所研究系统的固体溶液、低共熔混合物和共晶的形成。通常,低共熔混合物形成二元系统采用“V”形曲线,而共晶形成系统采用“W”形曲线表示两个共晶组分之间共晶的形成。有文献报道[75,76−78],可通过二元相图确定所研究的药物−共晶形成物组合的共晶形成区,其常见的二元相图如图10所示。
在溶液结晶过程中,由于各组分在溶剂体系中的溶解度不同,有时不能形成多组分共晶。因此,在尝试溶液结晶之前,需要确定包含三元组分的系统的热力学行为[80−81]。三元相图(API−CCF−溶剂相图)有助于确定在给定体系的共晶形成区域。许多研究人员利用三元相图作为优化工具来确定用于共晶形成的API 和共晶形成物的合适的共晶形成区[82−84]。当两个组分表现出相似的溶解度时,相图显示出更为对称的趋势。另一方面,当这两种组分表现出不同的溶解度时,相图的对称性就会减弱[85],如图11所示。
近年来,药物−药物共晶已经找到了临床前研究到批准的药品的方法。例如药物−药物共晶产品Entresto[86],在2015 年1 月获得了美国FDA 用于治疗慢性心力衰竭的批准,于2018年进入了重磅炸弹药物行列,全球销售额达到了10.28 亿美元。与构成该药物的两个单组分药物以及其他传统药物相比,Entresto在治疗心力衰竭方面显示出更好的效果,实现了一加一大于二的协同效应,导致心血管死亡人数减少20%,已成为治疗心力衰竭的首选药物[87]。由Mundipharma Research Limited Lab 研发的盐酸曲马多−塞来昔布共晶(Esteve)已经作为Ⅰ类创新药物进入中国市场(受理号:JXHL1700131)。在临床试验中,与盐酸曲马多、塞来昔布及两者组合相比,盐酸曲马多−塞来昔布共晶中API 的药代动力学参数得到了改善,其不仅可以有效降低盐酸曲马多的最大浓度、毒副作用和成瘾性,同时也可提高塞来昔布最大血药浓度,增加塞来昔布的镇痛作用,有效地缓解疼痛[88−89]。药物−药物共晶产品的上市为新药的研发提供了一种新的思路,有望缓解目前新药研发费时费力的局面,成为传统药品的可行替代品。
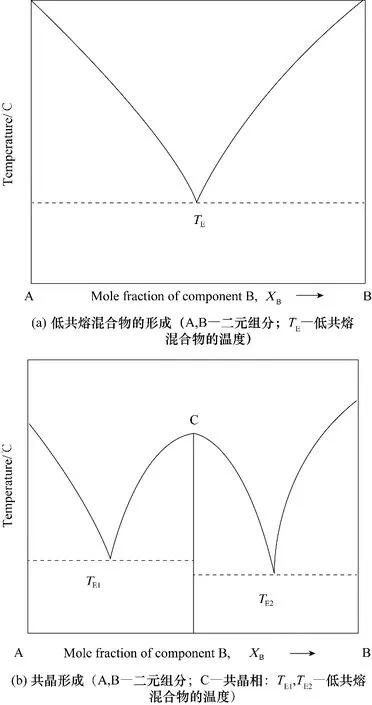
图10 二元相图[72,76−77,79]Fig.10 Schematic representation of binary phase diagram[72,76−77,79]
4 结论与展望

图11 三元相图(A—API;B—共晶形成物;C—溶剂;E1,E2—低共熔点)Fig.11 Schematic representation of ternary phase diagram(A—API;B—conformer;C—solvent;E1,E2—eutectic points)
药物−药物共晶的研发不仅在于改善原料药的物理化学性质,更重要的是将共晶药物作为新的药物联用方式,更大程度地提高药物的联用价值。本文从分子层面对药物−药物共晶的形成、溶解和代谢机理进行了详细阐述,并且重点介绍了如何进行药物−药物共晶设计和预测,为后续有关药物−药物共晶的研究提供指导。但目前药物−药物共晶的预测主要基于单个参数进行计算和预测,但共晶的形成受多种因素的影响,并且形成过程是复杂的,这极大地限制了共晶预测的准确性,开发可以预测药物−药物共晶的可靠模型将有助于预估药物−药物共晶的形成概率,为实验中共晶的筛选提供一定的理论参考价值。
尽管近几年药物−药物共晶的相关研究有了大幅增长,但大多都停留在对共晶结构的分析,在体内代谢的相关机理很少涉及。对于有特定化学计量比的药物−药物共晶,由于溶解性的不同,易溶的药物分子将会产生更高的毒副作用,这将大大限制药物−药物共晶的临床使用。如何设计共晶药物,开发老药新价值以及共晶药物如何满足药典和法律的规定等,还需要更多的关于共晶的研究数据来支撑。
猜你喜欢
中学生数理化·中考版(2022年12期)2022-02-16 07:37:00
建材发展导向(2021年14期)2021-08-23 00:57:14
中国煤层气(2019年2期)2019-08-27 00:59:30
模具制造(2019年3期)2019-06-06 02:11:04
环境与可持续发展(2017年2期)2017-04-06 03:07:30
含能材料(2017年1期)2017-03-04 15:46:20
含能材料(2017年7期)2017-03-04 11:16:26
中学化学(2016年10期)2017-01-07 08:47:24
当代化工研究(2016年6期)2016-03-20 16:21:48
中国学术期刊文摘(2016年8期)2016-02-13 13:04:44
